NPJ Vaccines. 2021 Feb 22;6(1):28.
doi: 10.1038/s41541-021-00292-w. PMID: 33619260.
https://www.nature.com/articles/s41541-021-00292-w
SARS-CoV-2 es un virus de ARN que pertenece a la familia Coronaviridae y causa la enfermedad COVID-19. El virus se habría originado en China, donde fue secuenciado inicialmente y se extendió rápidamente por todo el mundo mediante viajeros y turistas, convirtiéndose en una pandemia que, hasta el 5 de enero de 2021, causó más de 1.866.000 muertes. Una vacuna eficaz contra esta enfermedad será fundamental para reducir la morbilidad y la mortalidad.
El genoma del virus codifica varias proteínas estructurales y no estructurales, incluidas las proteínas Spike (S), de envoltura (E), de membrana (M) y nucleocápside (N). La mayoría de las vacunas candidatas para COVID-19 que emplean la administración de antígenos virales o secuencias de genes virales tienen como objetivo inducir anticuerpos neutralizantes contra la proteína Spike (S), previniendo la captación a través del receptor ACE2 humano y, por lo tanto, bloqueando la infección. Sin embargo, un número creciente de bibliografía ha destacado la importancia de las respuestas celulares en la recuperación de los pacientes con COVID-19, promoviendo así, no solo el uso de estrategias de vacuna que favorecen la inducción de respuestas mediadas por células T, sino también el screening de su producción en participantes de ensayos clínicos. Por otro lado, las estrategias que utilizan virus completo, ya sea atenuado o inactivado, aspiran a inducir una respuesta policlonal más amplia y heteróloga contra varios antígenos virales.
Desde la publicación de la secuencia del genoma del SARS-CoV-2, el 11 de enero de 2020, un esfuerzo de una velocidad y magnitud sin precedentes se propuso desarrollar una vacuna contra la enfermedad. Los avances recientes en el campo han hecho posible la emisión de autorizaciones de uso de emergencia (EUA) por parte de varias agencias reguladoras de medicamentos nacionales e internacionales para diferentes candidatos a vacunas contra el SARS-CoV-2 en menos de un año desde que se lanzó la secuencia del genoma del virus.
Una vacuna ideal contra el SARS-CoV-2 debe cumplir los siguientes requisitos: proteger no solo de enfermedades graves sino también contrarrestar la infección en todas las poblaciones vacunadas, incluidas las personas menos inmunodeprimidas; provocar respuestas inmunitarias de memoria a largo plazo después de un número mínimo de inmunizaciones o dosis de refuerzo; la empresa de fabricación debería poder aumentar la producción para producir miles de millones de dosis al año y tener el potencial de hacerla fácilmente accesible para las campañas de vacunación en todo el mundo a un costo asequible y en un tiempo limitado.
Cuatro iniciativas diferentes se encuentran entre las fuentes esenciales de financiación que permitieron el desarrollo de varias vacunas candidatas al SARS-CoV-2. Una de las primeras fuentes de financiación fue la Coalition for Epidemic Preparedness Innovations (CEPI), una asociación mundial sin fines de lucro que se dedica a proporcionar fondos para vacunas para detener las epidemias emergentes. Otra fuente vital de fondos provino de la Autoridad de Investigación y Desarrollo Biomédico Avanzado (BARDA), que ha asignado varios millones de dólares a los principales candidatos a vacunas y otros tratamientos prometedores de COVID-19. El programa de vacunas de la Unión Europea tiene en marcha un esfuerzo conjunto para comprar vacunas para los países de la UE. Esta entidad ya ha firmado contratos con seis desarrolladores de vacunas, incluidos Pfizer y BioNTech, Sanofi-GSK, Curevac, AstraZeneca y la Universidad de Oxford, Johnson & Johnson y Moderna. Más recientemente, la Operación Warp Speed del gobierno de EE. UU. invirtió más de mil millones de dólares para financiar el desarrollo de 8 candidatos de vacuna líderes para acelerar su evaluación, aprobación y fabricación para EE. UU. Por último, Gavi, una alianza de vacunas de acceso global, CEPI y la Organización Mundial de la Salud (OMS) han lanzado la iniciativa COVAX (Acceso a la vacuna contra el coronavirus) para garantizar el acceso equitativo de las vacunas del SARSCoV-2 a países no autofinanciados que carecen de recursos.
Según la OMS, al 5 de enero de 2021, hay 63 vacunas candidatas en ensayos clínicos en humanos y más de 172 candidatas en desarrollo preclínico en todo el mundo. Entre las 60 vacunas evaluadas clínicamente, se encontraron 13 candidatos líderes que ya están realizando o ingresando a ensayos clínicos de Fase 3. Las tecnologías de plataforma han sido empleadas por diferentes grupos de investigación para desarrollar sus candidatos a vacunas, en donde es de esperarse que los primeros candidatos a entrar en los ensayos clínicos de Fase 3 estén utilizando estrategias de despliegue rápido, a saber, plataformas de ácido nucleico, plataformas de vectores virales no replicantes, virus inactivados o vacunas de subunidades recombinantes. Otras estrategias tradicionales de desarrollo de vacunas, como las vacunas con virus atenuados, requieren largos procesos de cultivo celular para lograr cepas atenuadas. Es muy posible que las vacunas contra el SARS-CoV-2 de segunda generación demuestren la capacidad de provocar respuestas de memoria más sólidas y más largas con una sola inmunización.
Resumen de las estrategias utilizadas para el desarrollo y la administración de vacunas
Hay dos objetivos principales que cualquier estrategia de vacuna debe lograr: la seguridad de la vacuna y la producción de respuestas inmunitarias adaptativas sólidas que conducen a protección a largo plazo contra varias cepas del patógeno con -idealmente- una dosis de la vacuna.
Un creciente cuerpo de evidencia sugiere que las respuestas mediadas por células T contra el SARS-CoV-2 son extremadamente importantes y más duradera que la inmunidad de células B. Por lo tanto, las estrategias de vacuna que inducen fuertes respuestas celulares además de la inmunidad humoral presentan una ventaja significativa en la presente pandemia.
Es así, que existen varias estrategias (ver Figura 1):
A) Vacunas de patógenos atenuados: Las estrategias de vacuna de patógenos vivos atenuados consisten en administrar una forma debilitada de patógenos vivos. Los pases prolongados de cultivos celulares en líneas celulares no humanas o animales disminuyen la virulencia del patógeno. Este tipo de vacunas generalmente provoca respuestas inmunes de memoria robustas y de largo plazo después de una sola dosis.
B) Vacunas de patógenos inactivados: Las vacunas de patógenos inactivados contienen patógenos completos que han sido sometidos a inactivación por tratamiento térmico o químico.
C) Vacunas de subunidad: Las vacunas de la subunidad se preparan a partir de la purificación de antígenos de patógenos replicados en cultivos celulares o de antígenos expresados de forma recombinante. Estas vacunas comúnmente requieren la adición de adyuvantes para enviar señales de peligro a las células presentadoras de antígenos y provocar respuestas inmunes robustas.
D) Vacunas de partículas similares a virus: Las partículas similares a virus se pueden autoensamblar y liberar a partir de células de levadura recombinantes u otros sistemas de expresión como el sistema de expresión del virus vaccinia o incluso plantas de tabaco transfectadas con el virus del mosaico del tabaco.
E) Vacunas con vectores virales: Las vacunas de vectores virales utilizan una plataforma adenoviral o contra el sarampión manipulada genéticamente para expresar un antígeno extraño que comúnmente da como resultado una respuesta celular y humoral sólida.
F) y G) Vacunas de ácido nucleico: Las vacunas de ácido nucleico (ADN y ARNm) son muy rápidas de producir, pero no se probaron como estrategias exitosas de vacunas humanas. El ácido nucleico que codifica una proteína inmunogénica del patógeno, una vez administrado, es capturado por las células presentadoras de antígeno que lo utilizan para expresar y presentar el antígeno. Se prevé que estas vacunas tengan problemas menores de seguridad, ya que el ácido nucleico se degrada rápidamente dentro del cuerpo humano.
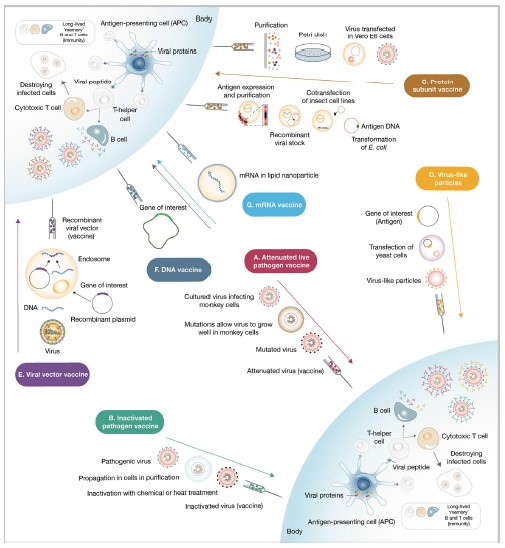
Fig 1: Resumen de las estrategias utilizadas para el desarrollo y la administración de vacunas
Candidatos actuales para la vacuna contra el SARS-CoV-2 en Fase 3 de la evaluación clínica
Vacunas de ácido nucleico
Vacunas de ARNm
ARNm-1273 (Moderna / US NIAID): Moderna Therapeutics, con sede en Boston, se asoció con el Instituto Nacional de Alergias y Enfermedades Infecciosas (NIAID) para producir la primera vacuna candidata que entró en ensayos clínicos en 63 días después de la secuenciación del genoma del SARS- CoV-2. La vacuna se basa en una molécula de ARNm que contiene la información para la síntesis de la forma de prefusión estabilizada de la proteína SARS-CoV-2 Spike (S) encapsulada en un vector de nanopartículas lipídicas (LNP) que mejora la captación por las células inmunes del huésped. El ARNm administrado utiliza la maquinaria de transcripción y traducción de la célula huésped para producir el antígeno viral que luego se presenta en los linfocitos T y también es reconocido directamente por los linfocitos B del huésped, iniciando así una respuesta inmune adaptativa dirigida contra la proteína del virus.
La dosificación de los primeros voluntarios con ARNm-1273 comenzó el 16 de marzo en 45 voluntarios sanos de entre 18 y 55 años de edad que recibieron tres dosis diferentes, a saber, 25 μg, 100 μg y 250 μg de ARN en forma de estimulación inicial. La segunda vacuna se administró 28 días después de la primera. El informe del ensayo de fase 1 describió una respuesta humoral dependiente de la dosis y la producción de anticuerpos neutralizantes en concentraciones similares a los detectados en sueros convalecientes de COVID-19. Además, las dos dosis más bajas provocaron una fuerte respuesta de los linfocitos T CD4 + con una expresión mínima de citocinas T helper 2 (Th2) que resultaron perjudiciales durante los esfuerzos de desarrollo de vacunas contra el SARS y el MERS (Síndrome respiratorio de Oriente Medio). Por otro lado, las respuestas de los linfocitos T CD8 + solo fueron provocadas por la dosis de 100 μg. El ARNm-1273 fue generalmente bien tolerado y no se informaron efectos adversos de la etapa 4 (incapacitante o potencialmente letal). Los eventos adversos, que comprendieron principalmente mialgia, fatiga, dolor de cabeza, escalofríos y dolor alrededor del lugar de la inyección, fueron más frecuentes después de la segunda inmunización y fueron más prominentes en el grupo de dosis alta (250 μg).
Recientemente se publicaron resultados adicionales de un pequeño estudio de fase 1 en 40 adultos mayores, que se dividieron en dos grupos de edad (56-70 años o ≥71 años). A los participantes se les administraron dos dosis de 25 μg o 100 μg de ARNm-1273 que previamente habían demostrado exhibir un perfil de tolerancia más alto. El perfil inmunogénico de ambos grupos de edad fue bastante similar al informado en el grupo de 18 a 55, lo que sugiere que esta estrategia de vacunación podría ser igualmente inmunogénica en los grupos de edad más vulnerables y generalmente menos inmunocompetentes. Se descubrió que la dosis de 100 μg era más inmunogénica, lo que respalda su uso en un ensayo de vacuna de Fase 3.
Después de completar un ensayo de vacunación de Fase 2 en 300 adultos jóvenes y 300 adultos mayores a los que se les administraron las dosis de 25 o 100 μg, el ARNm-1273 entró en un ensayo de eficacia de Fase 3 el 27 de julio (Identificador de ensayo clínico: NCT04470427). Este ensayo implica la inscripción de 30.000 participantes en los EE. UU., la mitad de los cuales recibirá 2 dosis de 100 μg de ARNm-1273 y la otra mitad un placebo en un estudio aleatorizado doble ciego controlado con placebo. El criterio de valoración principal del estudio es la prevención del COVID-19 sintomático con criterios de valoración secundarios que incluyen la prevención de la infección por SARS-CoV-2 y la prevención de la hospitalización por COVID-19.
El 18 de noviembre, Moderna anunció que su candidata a vacuna alcanzó su criterio de valoración principal de eficacia después de revisar el primer análisis intermedio de su estudio clínico de Fase 3. Este análisis inicial, establecido 14 días después de la segunda vacunación, incluye 95 casos confirmados de COVID-19 entre los participantes, 90 de los cuales pertenecían al grupo que recibió el placebo y 5 al grupo que recibió ARNm-1273. Los cálculos de división de casos revelan una eficacia inicial del 94,5% de la vacuna candidata. Otra observación importante fue que, de los 11 casos graves analizados, ninguno había ocurrido en el grupo vacunado con ARNm-1273, lo que sugiere que posiblemente el ARNm-1273 protege del COVID-19 grave, aunque se necesitan más datos para establecer la certeza. En cuanto al perfil de seguridad del ARNm-1273, se informaron principalmente eventos leves a moderados. Los efectos adversos graves más frecuentes fueron dolor en el lugar de la inyección después de la primera dosis (2,7%) y fatiga (9,7%), mialgia (8,9%), artralgia (5,2%), dolor de cabeza (4,5%), dolor (4,1%). y enrojecimiento en el lugar de la inyección (2,0%) después de la segunda dosis. En general, estos efectos se describieron como de corta duración.
En un segundo comunicado de prensa nuevamente el 16 de noviembre, Moderna reveló que después de pruebas adicionales, se encontró que su vacuna candidata se mantuvo estable durante 30 días entre 2 ° y 8 ° C, hasta por 6 meses a -20 ° C y por hasta 12 h a temperatura ambiente, abordando así uno de los mayores desafíos que presentan las vacunas de ARNm que es la logística de su distribución en áreas rurales y la necesidad de refrigeradores especializados.
El 30 de noviembre, Moderna anunció los resultados del análisis de eficacia principal de su estudio de Fase 3 que involucra 196 casos de COVID-19 confirmado. Entre los 30 individuos que desarrollaron una enfermedad grave, ninguno había sido inmunizado con la vacuna candidata, lo que sugiere que el ARNm-1273 protege fuertemente contra la enfermedad grave. Además, de los 196 casos confirmados de COVID-19, solo 11 pertenecían al brazo de ARNm-1273 del estudio, lo que arroja una estimación puntual de la eficacia de la vacuna del 94,1% 2 semanas después de la segunda dosis. Se informó que la eficacia era constante en todas las edades, razas y etnias, mientras que no se informaron diferencias en el perfil de seguridad del candidato publicado anteriormente. Por lo tanto, la compañía alcanzó los dos puntos finales de 151 casos confirmados y un seguimiento promedio de dos meses de los participantes establecidos en el diseño del estudio y el 18 de diciembre, la Administración de Alimentos y Medicamentos (FDA) de EE. UU. otorgó la EUA de la vacuna en individuos mayores de 18 años de edad, seguido Autoridad Sanitaria de Canadá el 23 de diciembre. El 30 de diciembre, los resultados de seguridad y eficacia del ensayo de Fase 3 del ARNm-1273 se publicaron en el New England Journal of Medicine, confirmando el perfil de seguridad y eficacia del 94,1% de la vacuna candidata. El 4 de enero de 2021, Israel también aprobó la vacuna candidata de Moderna y, posteriormente, la Agencia Europea de Medicamentos (EMA) recomendó la autorización de la vacuna.
ARNm-BNT162b2 / Comirnaty (Pfizer / BioNTech / Fosun Pharma): La segunda plataforma de ARNm candidata es BNT162b2 desarrollada por Pfizer en colaboración con BioNTech (abreviatura de Nuevas Tecnologías Biofarmacéuticas) con sede en Alemania y Fosun Pharma, con sede en Shangai. BioNTech desarrolló y probó inicialmente cuatro candidatos a vacunas modificadas basadas en ARNm (modRNA) diseñadas para administrarse en dos vacunaciones con 3 semanas de diferencia y, tras la inserción en el citoplasma de la célula huésped, instruye a las células inmunitarias para que realicen varias copias de la proteína Spike de SARS-CoV-2 de longitud completa.
Los resultados de los ensayos clínicos aleatorizados controlados con placebo de Fase 1 mostraron que BNT162b2 genera efectos secundarios mínimos tanto en participantes más jóvenes (18-55 años) como mayores (65-85 años). Además, se evaluaron dos candidatos diferentes en estos ensayos, a saber, BNT162b1 y BNT162b2. Ambos candidatos indujeron la producción de concentraciones de anticuerpos neutralizantes dependientes de las dosis igualmente altas contra el SARS-CoV-2 en los participantes inoculados. De hecho, las concentraciones de anticuerpos neutralizantes fueron superiores o iguales a los de los sueros convalecientes del SARS-CoV-2. Sin embargo, BNT162b2 demostró menos reactogenicidad sistémica en adultos mayores y, por lo tanto, la dosis de 30 μg de este candidato fue seleccionada para estudios de eficacia de Fase 2/3 a gran escala. Las respuestas de linfocitos T no se informaron inicialmente para este candidato específico, pero en base a un estudio previo sobre la inmunogenicidad de BNT162b1 se espera una activación significativa de células T CD8 + específicas y poblaciones sesgadas de Th1 CD4 +. De hecho, se descubrió que una inmunización de dos dosis con 30 µg / dosis de BNT162b1 inducía concentraciones elevadas de anticuerpos neutralizantes anti-SARS-CoV-2 y respuestas de linfocitos T Th1 y CD8 + específicas para el paciente. Se realizarán ensayos controlados con placebo aleatorizados de seguridad y eficacia de Fase 2/3 en 43488 voluntarios, incluidos individuos con afecciones crónicas subyacentes y diferentes antecedentes genéticos (Identificador de ensayo clínico: NCT04368728).
El criterio de valoración principal del ensayo es la prevención de COVID-19, y los criterios de valoración secundarios incluyen la prevención de COVID-19 grave y la prevención de la infección por SARS-CoV-2. El ensayo está diseñado para llevarse a cabo en 166 sitios de investigación clínica en todo el mundo en al menos 3 continentes diferentes. La capacidad de fabricación de Pfizer permitirá un suministro global de hasta 50 millones de dosis para fines de 2020 y 1.300 millones de dosis para fines de 2021 si su candidata a vacuna logra la autorización. En un comunicado de prensa emitido el 18 de noviembre, Pfizer y BioNTech anunciaron que los datos del análisis intermedio final sugieren que su candidato demostró una eficacia del 95% contra COVID-19 una semana después de administrar ambas inmunizaciones. Estos datos se basan en la evaluación de 43.448 participantes, 170 de los cuales desarrollaron COVID-19 en la ventana de evaluación. Entre ellos, 162 pertenecen al grupo placebo y 8 al grupo que fue inmunizado con la vacuna candidata. Según el comunicado de prensa, la eficacia de la vacuna fue constante según la edad, el sexo, la raza y la etnia. Además, la eficacia observada en participantes vacunados mayores de 65 años fue superior al 94%. Con respecto al perfil de seguridad de BNT162b2, la vacuna candidata fue bien tolerada en todas las poblaciones ya que no se informaron problemas de seguridad graves. Los eventos de Grado 3 más frecuentes fueron fatiga presentada en 3.8% y dolor de cabeza en 2.0% de los participantes vacunados. Según estos resultados, la empresa considera que se ha cumplido el hito de datos de seguridad exigido por la FDA de EE. UU. para la EUA y el 20 de noviembre, Pfizer y BioNtech se convirtieron en las primeras empresas que presentaron una solicitud de EUA de una vacuna contra el SARS-CoV-2 a la FDA. El 2 de diciembre, BNT162b2 recibió la EUA de la Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA), el regulador de medicamentos del Reino Unido y la administración de vacunas comenzó el 8 de diciembre en el Reino Unido, seguido de una autorización otorgada por la agencia reguladora de medicamentos canadiense el 9 de diciembre. El 11 de diciembre, la FDA otorgó la EUA para BNT162b2 en individuos mayores de 16 años, mientras que la EMA y la Comisión Europea aprobaron la vacuna para individuos mayores de 16 años que residan en cualquiera de los 27 estados miembros de la UE el 21 de diciembre.
Desde entonces, varios otros países han otorgado la EUA para BNT162b2, entre ellos Argentina, Chile, Ecuador, Costa Rica, México, Panamá, Kuwait y Singapur. Suiza, Bahréin y Arabia Saudita también han autorizado la vacuna, y el 31 de diciembre la OMS otorgó la validación de emergencia a BNT162b2, permitiendo así a varios países acelerar los procesos de autorización, importación y distribución de este candidato.
El mismo día se publicaron los resultados de seguridad y eficacia del ensayo clínico BNT162b2 Fase 3 en el New England Journal of Medicine. Además de la confirmación de los 8 casos de COVID-19 entre los participantes asignados a recibir BNT162b2 y 162 casos entre los asignados al placebo se reveló un hallazgo adicional; en 10 casos notificados de COVID-19 grave que se manifestaron después de que los participantes habían recibido la primera dosis, 9 ocurrieron en el grupo de placebo y solo 1 en un receptor de BNT162b2, lo que sugiere que BNT162b2 protege adicionalmente del COVID-19 grave.
Vacunas de ADN
INO-4800 (Inovio / International Vaccine Institute): Aunque la empresa Inovio, con sede en Pensilvania, aún no ha entrado oficialmente en los ensayos de Fase 3, su candidata es la vacuna de ADN del SARS-CoV-2 más avanzada hasta el momento. Inovio Pharmaceuticals ha desarrollado varias vacunas experimentales basadas en ADN que se administran por vía intradérmica con la ayuda de un dispositivo portátil llamado «Cellectra 2000» que emite un pequeño pulso eléctrico que permite una absorción celular y nuclear eficiente de las moléculas de ADN a través de un mecanismo de electroporación. Su candidato es una vacuna de dos dosis.
El 30 de junio, un anuncio de la compañía reveló datos provisionales de un ensayo de Fase 1 en 36 voluntarios de 18 a 50 años de edad que recibieron dos dosis de 1.0 mg o 2.0 mg de INO-4800 con cuatro semanas de diferencia. Estos resultados se publicaron el 23 de diciembre y, según el artículo, no se informaron efectos adversos graves y 34 de los 36 participantes presentaron fuertes respuestas humorales en los grupos de 1,0 mg y 2,0 mg. Además, el 78% de los participantes en el brazo de 1,0 mg y el 84% de los participantes en el brazo de 2,0 mg generaron anticuerpos neutralizantes según lo evaluado por un ensayo de neutralización de cepas SARS-CoV-2 / Australia / VIC01 / 2020. Finalmente, el 74% y el 100% de los participantes inmunizados con 1,0 mg y 2,0 mg, respectivamente, desarrollaron fuertes respuestas de células T Th1 y CD8 +.
El 28 de septiembre, Inovio anunció que la FDA había suspendido parcialmente los ensayos clínicos en Fase 2/3 planificados de la vacuna candidata debido a preguntas sobre el diseño y uso de ‘Cellectra 2000’. El 16 de noviembre, Inovio dijo que el FDA les había dado permiso para seguir adelante con su ensayo de Fase 2/3 llamado INNOVATE65 (Identificador de ensayo clínico: NCT04642638). El brazo de Fase 2 de este estudio se llevará a cabo con 400 voluntarios que recibirán dos dosis por vía intradérmica de 1.0 o 2.0 mg de INO-4800 o un placebo con un intervalo de 4 semanas. El segmento de Fase 3 del estudio involucrará a 6.178 voluntarios que recibirán dosis determinadas por los resultados de seguridad e inmunogenicidad obtenidos del segmento de Fase 2.
Vacunas de vectores virales con replicación defectuosa
Ad5-nCoV (CanSino Biological / Instituto de Biotecnología de Beijing / Academia de Ciencias Médicas Militares): La empresa china CanSino Biologics, en colaboración con el Instituto de Biología de la Academia de Ciencias Médicas Militares de China, desarrolló un candidato que utiliza el vector de adenovirus humano serotipo 5 (Ad5) para entregar la información que codifica la proteína S de longitud completa del SARS-CoV-2 en las células huésped. Ad5 es el principal serotipo adenoviral en humanos, lo que significa que un porcentaje significativo de individuos puede tener contacto reciente y, por lo tanto, inmunidad preexistente contra el vector viral que también podría obstaculizar respuestas inmunes robustas contra el antígeno presentado.
Los datos preliminares de seguridad e inmunogenicidad de Fase 1 obtenidos de 108 participantes entre 18 y 60 años que recibieron dosis bajas, medias y altas de Ad5-nCoV se publicaron el 22 de mayo. Se encontró que las dos dosis más bajas de 5 × 1010 y 1 × 1011 partículas virales tenían un perfil de inmunogenicidad y seguridad aceptable y se seleccionaron para un ensayo de Fase 2.
Los resultados del ensayo de Fase 2, doble ciego, aleatorizado y controlado con placebo también se publicaron el 20 de julio. Se aplicó cualquiera de las dos dosis seleccionadas de Ad5-nCoV o el placebo a un total de 508 voluntarios elegibles de 18 a 83 años de edad. Ambos grupos de dosis provocaron anticuerpos anti-RBD en más del 95% de los participantes el día 28 después de la inmunización y concentraciones de anticuerpos neutralizantes contra el SARS-CoV-2 vivo. Además, alrededor del 90% de los participantes vacunados de ambos grupos demostraron activación de respuestas específicas de células T como lo demuestra el ensayo ELISpot de interferón-γ. El 72% y el 74% de los participantes en los grupos de dosis más alta y más baja informaron reacciones adversas leves, respectivamente. Se documentaron reacciones adversas graves en menos del 10% de los participantes de cada grupo y no se informaron reacciones adversas serias. A raíz de estos resultados, se seleccionó el régimen de una única inmunización de 5 × 1010 partículas virales para proceder en los ensayos de eficacia de Fase 3 que evalúan la protección contra la incidencia de COVID-19 grave con la inscripción de 40.000 voluntarios en Arabia Saudita, Rusia y Pakistán. (Identificador de ensayo clínico: NCT04526990). Mientras tanto, el 25 de junio, la Comisión Militar Central de China anunció que Ad5-nCoV recibió la aprobación para su uso en la adquisición y análisis militar en ausencia de los resultados del ensayo de Fase 3.
AZD1222 (AstraZeneca / Universidad de Oxford): La vacuna vectorizada viral de la Universidad de Oxford y AstraZeneca representa otro candidato. Fue uno de los primeros en comenzar los ensayos clínicos y el único que utilizó una plataforma debilitada de adenovirus de chimpancé (ChAdOx1) para eludir el problema de la inmunidad preexistente contra el vector, ya que muy pocos humanos, si es que hay alguno, tendrían un contacto previo con un virus simio. El vector ChAdOx1 se ha diseñado para incluir la información que codifica la proteína Spike del SARS-CoV-2 de tipo salvaje.
Los primeros resultados de un estudio de control multicéntrico, aleatorizado, simple ciego de fase 1/2 de 1090 voluntarios adultos sanos de entre 18 y 55 años se informaron el 20 de julio. Una proteína meningocócica comparada con una vacuna conjugada meningocócica (MenACWY) sirvió como control en este estudio. Los participantes recibieron una o dos dosis que contenían 5 × 10(10) partículas virales con 4 semanas de diferencia o la vacuna meningocócica de control. El perfil de seguridad de la vacuna se caracterizó como aceptable, mientras que la vacunación homóloga provocó respuestas neutralizantes contra el SARS-CoV-2 en todos los participantes, pero no tuvo efectos adicionales sobre las respuestas adaptativas celulares.
Se están llevando a cabo ensayos de Fase 3 de eficacia y seguridad con el régimen de dos dosis (Identificador de ensayo clínico: NCT04516746) en más de 30.000 personas en los EE. UU., India (Identificador de ensayo clínico: CTRI / 2020/08/027170), Brasil (Identificador del ensayo: ISRCTN89951424), Rusia (Identificador del ensayo clínico: NCT04540393) y Sudáfrica. El 6 de septiembre, un evento adverso grave (AAG) presentado en un paciente inscrito en estudios de Fase 3 detuvo temporalmente los ensayos que se reanudaron en la mayoría de los países, incluido el Reino Unido, pero no en los EE. UU. Esa fue la segunda pausa temporal para un estudio AZD1222, ya que en julio el ensayo se detuvo durante varios días después de que un participante desarrollara síntomas neurológicos graves. Posteriormente se concluyó que estos síntomas se debían a un caso de esclerosis múltiple no diagnosticado previamente que no estaba relacionado con la vacuna. Los criterios de valoración primarios del estudio de Fase 3 se centran en la prevención de COVID-19 y el perfil de reactogenicidad y tolerancia del candidato.
El 23 de noviembre, la Universidad de Oxford y AstraZeneca emitieron comunicados de prensa separados presentando los resultados provisionales de la Fase 3 de su vacuna candidata. Estos resultados se publicaron el 8 de diciembre en Lancet en un análisis de seguridad y eficacia de cuatro ensayos llevados a cabo en Brasil, Sudáfrica y el Reino Unido, lo que representa poblaciones geográfica y étnicamente diversas. El análisis incluyó 131 casos confirmados de COVID-19 detectados en dos regímenes de dosificación diferentes, incluidos 11,636 participantes reclutados en el brazo inglés y brasileño del estudio. El primer régimen se aplicó dos dosis completas con 4 semanas de diferencia en 8895 participantes adultos y mostró una eficacia del 62,1%. El segundo régimen, que se reconoció como el resultado de un error logístico, implicó la mitad de la primera dosis seguida de una dosis de refuerzo completa con la misma separación cronológica entre ellos e incluyó a 2741 individuos de 18 a 55 años que mostraban un 90 % eficacia. Se plantea la hipótesis de que esta discrepancia en la eficacia podría deberse a la combinación del grupo de edad más joven de la cohorte más pequeña y al hecho de que una dosis inicial más alta podría promover la inducción de anticuerpos contra el vector viral, dificultando así la intensidad de las respuestas inmunitarias inducidas por la dosis de refuerzo. La eficacia combinada de toda la cohorte de participantes 14 días después de la segunda dosis fue del 70,4% sin que se informaran casos graves u hospitalizaciones entre los participantes vacunados. Además, 3 semanas después de la administración de la primera dosis se reportaron diez casos de hospitalización por COVID-19, todos en el grupo control. Los datos preliminares sugieren que AZD1222 podría reducir la transmisión del virus ya que se observó una reducción en las infecciones asintomáticas. En el brazo de eficacia, se informa que el perfil de seguridad de la vacuna candidata es generalmente bueno y que las reacciones adversas son menos intensas y frecuentes en los participantes inmunizados de mayor edad que recibieron dosis más bajas y estos eventos disminuyen después de la segunda dosis.
AstraZeneca, con el apoyo de la Universidad de Oxford, presentó la seguridad y eficacia de la Fase 3 provisional completa a varios reguladores, incluidos el Reino Unido, la EMA y Brasil para su revisión y aprobación de uso de emergencia de su candidato. El 27 de noviembre, la MHRA emitió un comunicado de prensa informando que el Departamento de Salud y Asistencia Social del Reino Unido solicitó oficialmente la revisión de la vacuna candidata AZD1222 y el 30 de diciembre recibió la EUA para personas mayores de 18 años en el Reino Unido y Argentina. El 3 de enero de 2021, India otorgó la EUA a AZD1222, mientras que el 4 de enero comenzaron las primeras vacunas con AZD1222 en el Hospital Churchill de Oxford.
Gam-COVID-Vac / Sputnik V (Instituto de Investigación Gamaleya / Ministerio de Salud de la Federación de Rusia / Investigación y desarrollo de fármacos contractuales de Acellena): Los científicos del Instituto de Investigación Ruso Gamaleya desarrollaron la única vacuna candidata heteróloga de refuerzo primario del SARSCoV-2 hasta el momento con el fin de sortear el desafío de la inmunogenicidad reducida debido a los anticuerpos producidos contra el vector viral después de la primera inmunización. El serotipo del vector adenoviral utilizado para la vacunación de cebado es diferente del serotipo adenoviral utilizado como refuerzo. Por lo tanto, se seleccionó Ad26 con replicación defectuosa para entregar la información genética de la proteína Spike durante la primera vacunación y Ad5 recombinante con replicación defectuosa durante la segunda.
La vacuna candidata, recientemente rebautizada como Sputnik V, se probó en dos ensayos de Fase 1/2 a pequeña escala en los que participaron 38 participantes cada uno. Los resultados de los dos estudios se publicaron el 4 de septiembre, 3 semanas después de que el presidente Putin anunciara la autorización del Sputnik V para un uso limitado. El informe del ensayo de Fase 1/2 describió un buen perfil de seguridad con efectos adversos leves en una parte de los participantes vacunados, como astenia, mialgia, artralgia, fiebre, dolor de cabeza y dolor en el lugar de la inyección. El perfil inmunogénico del candidato a vacuna también fue bueno al inducir fuertes respuestas humorales en todos los participantes, así como la activación de las células T CD4 + y CD8 +.
El hecho de que el Ministerio de Salud de la Federación de Rusia haya aprobado Sputnik V como la primera vacuna para COVID-19 antes de los ensayos de seguridad y eficacia de la Fase 3 ha causado controversia y preocupación en la comunidad científica. Los ensayos de Fase 3 se planearon inicialmente para solo 2000 voluntarios, pero luego se reprogramaron para inscribir a 40,000 personas en 45 centros médicos diferentes en Rusia (Identificador de ensayo clínico: NCT04530396) y la República de Bielorrusia (Identificador de ensayo clínico: NCT04564716). El criterio de valoración principal es demostrar que Sputnik V evita que los participantes vacunados desarrollen COVID-19.
El 24 de noviembre, los desarrolladores de Sputnik V anunciaron los resultados de su segundo informe provisional de Fase 3 que reveló una eficacia del 91,4% de esta vacuna candidata después de analizar los datos obtenidos de 18.794 personas una semana después de recibir ambas dosis del régimen de inmunización heterólogo. Este análisis se basa en 39 casos confirmados de COVID-19 entre los participantes, de los cuales se informó que 8 pertenecían al grupo vacunado y 31 al grupo placebo. Según el comunicado de prensa, no se detectaron eventos adversos potencialmente mortales (Grado 4), mientras que los eventos graves más comunes (Grado 3) reportados fueron dolor en el lugar de la inyección y síntomas similares a los de la gripe como fiebre, fatiga y dolor de cabeza.
El 14 de diciembre, el Centro Nacional de Gamaleya y uno de sus patrocinadores, el Fondo Ruso de Inversión Directa (RDIF), anunciaron los resultados de su análisis intermedio final luego de llegar al punto de control de 78 casos confirmados de COVID-19. Se informó que la eficacia de Sputnik V tres semanas después de la administración de la primera dosis fue del 91,4% según el análisis de los datos obtenidos de 22.714 participantes (17.032 recibieron la vacuna y 5682). En el brazo inmunizado se notificaron 16 casos de COVID-19 frente a 62 casos en el brazo de placebo. Además, el anuncio informó 20 casos graves de COVID-19 en el grupo de placebo y ningún caso de enfermedad grave en el grupo vacunado, afirmando que la vacuna demostró una eficacia del 100% contra COVID-19 grave. Además de la aprobación de uso temprano de Sputnik V otorgada en Rusia, también se han emitido la EUA para esta vacuna desde Bielorrusia y Argentina.
JNJ-78436735 / Ad26.COV2.S (Centro Médico Janssen y Beth Israel Deaconess): Janssen Pharmaceuticals es la rama de desarrollo de vacunas de Johnson & Johnson Pharmaceutic. Su candidato es un vector basado en adenovirus 26 con replicación defectuosa que expresa la proteína S estabilizada previa a la fusión del SARS-CoV-2, un método desarrollado hace una década por investigadores del Beth Israel Deaconess Medical Center en Boston. Su principal diferencia con la vacuna candidata CanSino es el serotipo de adenovirus. A diferencia del omnipresente serotipo Ad5, muy pocas personas han estado expuestas al raro serotipo Ad26, por lo tanto, no se espera que la inmunidad preexistente contra el vector que reduce la inmunogenicidad de este candidato sea una preocupación importante. La segunda ventaja de este candidato es que el esquema de dosificación implica una única inmunización.
Janssen lanzó en julio un ensayo multicéntrico de Fase 1/2, aleatorizado, doble ciego y controlado con placebo, cuyos resultados se informaron recientemente. JNJ-78436735 se administró a niveles de dosis de 0,5 × 10(11) o 1 × 10(11) partículas virales por vacunación en participantes de dos grupos de edad diferentes: el primer grupo estaba compuesto por 402 adultos sanos de entre 18 y 55 años y el segundo grupo estaba compuesto por 394 ancianos sanos de 65 años o más.
La reactogenicidad de la vacuna fue leve y causó principalmente dolor en el lugar de la inyección, fiebre, dolor de cabeza y mialgia. Se detectaron anticuerpos específicos contra la proteína S en el 92% de los participantes del grupo más joven que recibieron cualquiera de las dosis y alcanzaron una tasa de seroconversión del 100% en el grupo de mayor edad. Se observaron respuestas de células T CD4 + en más del 80% de los miembros de cualquier grupo y también se documentaron respuestas robustas de células T CD8 +.
Estos resultados llevaron a un ensayo de Fase 3 que reclutó hasta 60.000 participantes que recibirán el nivel de dosis de 0,5 × 10(11) partículas virales (Identificador de ensayo clínico: NCT04505722). Los resultados primarios del ensayo analizados fueron la aparición de COVID-19 de moderado a grave. El 12 de octubre, los ensayos clínicos de Fase 3 de Janssen se detuvieron después de que un participante del estudio manifestara una reacción adversa seria (SAE). No se han publicado datos sobre cuál era la naturaleza de la enfermedad o si el participante había recibido la vacuna candidata o el placebo, pero el 23 de octubre, Jannsen anunció que el ensayo de Fase 3 de su candidata estaba a punto de reanudarse después de la recomendación de la Junta de Monitoreo y Seguridad de Datos (DSMB) que supervisa el estudio.
El 15 de noviembre, Janssen informó que iniciarán un segundo ensayo clínico de Fase 3, aleatorizado, doble ciego y controlado con placebo que estudia la seguridad y eficacia de un régimen de dos dosis de su candidato (Identificador de ensayo clínico: NCT04614948). En el estudio participarán 30.000 adultos de Bélgica, Colombia, Francia, Alemania, Filipinas, Sudáfrica, España, Reino Unido y Estados Unidos que recibirán dos dosis de la vacuna candidata Ad26.COV2.S o un placebo con un intervalo de 57 días.
Vacunas de patógenos inactivados
CoronaVac (Sinovac Research and Development Co.): Lanzada inicialmente con el nombre PiCoVacc, CoronaVac, es una vacuna candidata con adyuvante de alumbre de virus inactivado y purificado. El candidato fue producido por la activación de β-propiolactona de la cepa CN2 de SARS-CoV-2 aislada del lavado broncoalveolar de un paciente hospitalizado, esta cepa está estrechamente relacionada con la cepa 2019-nCoV-BetaCoV Wuhan / WIV04 / 2019. Las vacunas inactivadas presentan algunos desafíos técnicos como desventaja. El proceso de inactivación a veces puede dañar los antígenos y provocar una inmunogenicidad subóptima. Además, las vacunas inactivadas comúnmente necesitan varias dosis de refuerzo para producir respuestas inmunes fuertes y no activan tradicionalmente las respuestas celulares. Más importante aún, para mejorar su capacidad de provocar inmunidad, estas vacunas requieren la adición de adyuvantes. De hecho, CoronaVac es una vacuna candidata con adyuvante de alumbre.
Se llevó a cabo un ensayo de inmunogenicidad y seguridad de escalada de dosis, aleatorizado, doble ciego y controlado con placebo de Fase 2 en el que participaron 600 voluntarios sanos de entre 18 y 59 años que recibieron dos dosis diferentes de la vacuna (3 o 6 μg / 0,5 ml) o placebo. CoronaVac fue bien tolerado en ambas dosis y la mayoría de las reacciones adversas fueron leves. El dolor en el lugar de la inyección fue el síntoma más común de los reportados. Ambas dosis de CoronaVac indujeron una seroconversión en más del 90% de los individuos inmunizados, sin embargo, no se informaron respuestas de células T.
CoronaVac se encuentra actualmente en ensayos clínicos de Fase 3 en un régimen de inyección de dos dosis con un intervalo de 14 días. Estos ensayos inscribirán a 8870 participantes de Brasil (Identificador de ensayo clínico: NCT04456595), voluntarios de Indonesia (Número de registro: INA-WXFM0YX) y Turquía, para evaluar la eficacia de la vacuna en la prevención del COVID-19 y la frecuencia de efectos adversos.
Según se informa, CoronaVac recibió una aprobación de emergencia para uso limitado en julio en China, mientras que Sinovac está aumentando la producción de su vacuna candidata con el objetivo de suministrar a Indonesia 40 millones de dosis para marzo de 2021 y comenzar la distribución mundial a principios de 2021.
Nombre desconocido (Instituto de Productos Biológicos de Wuhan / Grupo Nacional de Biotecnología de China-Sinopharm): El Instituto de Productos Biológicos de Wuhan se asoció con la farmacéutica estatal Sinopharm para desarrollar una vacuna de virus inactivado y purificado que se envió a los ensayos clínicos de Fase 1 y Fase 2. Este candidato se desarrolló al aislar inicialmente la cepa WIV04 de SARS-CoV-2 de un paciente en el Hospital Jinyintan, Wuhan. A continuación, el virus se propagó en una línea celular Vero y se inactivó con β-propiolactona. Finalmente, la vacuna se sometió a un procedimiento de adsorción de adyuvante de alumbre.
El informe provisional de los ensayos de Fase 1 y Fase 2 sobre adultos sanos entre 18 y 59 años se publicó el 13 de agosto. En el ensayo de dosis y seguridad de Fase 1, 96 participantes recibieron una de tres dosis diferentes (2,5, 5 y 10 μg / dosis) o un control de adyuvante de alumbre solo en un régimen de tres inyecciones. En el ensayo de seguridad e inmunogenicidad de Fase 2, se asignó aleatoriamente a 224 adultos para recibir dos veces una inmunización de 5 μg / dosis con un intervalo de 2 o 3 semanas entre cada dosis o recibir el control de adyuvante solo. Los resultados mostraron que la vacuna candidata tenía un buen perfil de seguridad con solo efectos adversos leves documentados (principalmente dolor en el lugar de la inyección y fiebre), producía anticuerpos en voluntarios, algunos de los cuales experimentaron fiebre y otros efectos secundarios. Ambos grupos que recibieron la vacuna candidata produjeron concentraciones altas de anticuerpos neutralizantes contra el SARS-CoV-2, aunque fueron significativamente más altas en el grupo que recibió las dos inyecciones con 3 semanas de diferencia.
Actualmente se están llevando a cabo ensayos clínicos de Fase 3 en los Emiratos Árabes Unidos (número de registro: ChiCTR2000034780), Perú, Marruecos (número de registro: ChiCTR2000039000) y Bahrein.
BBIBP-CorV (Instituto de Biotecnología de Beijing / Grupo Nacional de Biotecnología de China-Sinopharm): El segundo candidato a vacuna de virus inactivado desarrollado por Sinopharm es el resultado de su colaboración con el Instituto de Productos Biológicos de Beijing. BBIBP-CorV se desarrolló mediante la inactivación mediada por β-propiolactona de la cepa SARSCoV-2 19nCoV-CDC-Tan-HB02 que se replicó en células Vero88 y se adyuvó con hidróxido de aluminio. El hidróxido de aluminio activa la subunidad del receptor NLRP3 del inflamasoma y promueve la secreción de altos niveles de IL-1β e IL-18 derivadas del inflamasoma, activando así los mecanismos proinflamatorios del sistema inmunológico.
En el estudio de Fase 1 de seguridad y aumento de dosis, 192 participantes recibieron 2 μg, 4 μg u 8 μg de la vacuna o el placebo. Se emplearon dos grupos de edad, a saber, 18 a 59 años y ≥ 60 años. El efecto adverso sistemático más común informado fue fiebre en menos del 10% de los candidatos. El perfil de seguridad de la vacuna fue bastante bueno ya que todas las reacciones adversas documentadas fueron leves o moderadas y no se informaron eventos graves. La inmunogenicidad del candidato fue mayor en el grupo de edad más joven (18-59 años) y las concentraciones de anticuerpos neutralizantes presentaron una inducción dependiente de la dosis.
En los ensayos de Fase 2, se reclutaron 448 voluntarios que recibieron una dosis de 8 μg o dos dosis de 4 μg de vacuna con 2, 3 o 4 semanas de diferencia. Nuevamente, las reacciones adversas notificadas fueron leves o moderadas y la reacción sistemática más frecuente fue fiebre en menos del 4% de los miembros de cada grupo de dosis. Las concentraciones de anticuerpos neutralizantes fueron significativamente más altas en los grupos que recibieron una inmunización de refuerzo con 4 μg / dosis y fueron más altas cuando las dos inmunizaciones estaban separadas por una distancia de 3 semanas. Como era de esperar, no se informaron respuestas inmunitarias celulares. En este momento BBIBP-CorV se encuentra en ensayos clínicos de Fase 3 en Argentina (Identificador de ensayo clínico: NCT04560881), Bahrein, Jordania, Egipto y Emiratos Árabes Unidos (Identificador de ensayo clínico: NCT04510207), (Número de registro: ChiCTR2000034780) que evalúa la incidencia de COVID-19 en personas que han recibido dos dosis de la vacuna.
Según los informes, las dos candidatas a vacunas de Sinopharm están programadas para estar listas para el mercado a finales de año. El 14 de septiembre, U.A.E. otorgó aprobación de emergencia para que las vacunas de Sinopharms se administraran a los trabajadores de la salud, antes de obtener datos a gran escala sobre su seguridad y eficacia. El 9 de diciembre, U.A.E. otorgó la aprobación total a BBIBP-CorV, seguida de Bahrein el 13 de diciembre. El 30 de diciembre, Sinopharm anunció que su candidato tenía una eficacia del 79,34% y recibió la aprobación en China al día siguiente. Por otro lado, Egipto emitió una autorización de emergencia el 3 de enero de 2021.
Covaxin / BBV152 (Bharat Biotech / Consejo Indio de Investigación Médica / Instituto Nacional de Virología): Bharat Biotech, con sede en India, y el Consejo Indio de Investigación Médica desarrollaron un candidato a vacuna de virión completo inactivado purificado llamado Covaxin. La vacuna se desarrolló mediante la inactivación con β-propiolactona de una cepa india del nuevo coronavirus aislada por el Instituto Nacional de Virología de la India y propagada en células Vero CCL-81.
El 15 de diciembre de 1999 se publicó una preimpresión de un ensayo clínico de Fase 1 de BBV152. Los autores informan los resultados obtenidos de 375 participantes que recibieron tres formulaciones diferentes de BBV152 (n = 100 por cada formulación) o el adyuvante Algel (a base de aluminio) (n = 75). Las tres formulaciones diferentes incluían 3 o 6 μg de SARS-CoV-2 inactivado con virión completo adsorbido en alumbre (Algel-imidazoquinolina) o 6 μg de SARSCoV-2 inactivado con virión completo adsorbido en Algel solo. Los participantes recibieron dos dosis intramusculares con 2 semanas de diferencia y se evaluó la seguridad e inmunogenicidad de cada formulación. Se encontró que los efectos adversos fueron leves o moderados con una tasa de incidencia entre el 10 y el 20% y el dolor en el lugar de la inyección fue el evento informado con mayor frecuencia. En ambos grupos de Algel-imidazoquinolina, se informó la inducción de concentraciones altas de anticuerpos anti-SARS-CoV-2, células T CD4 + y CD8 + y fueron significativamente más altas que en el grupo de solo Algel. Las respuestas de las células T parecían estar sesgadas por Th1, mientras que las tasas de seroconversión después de la segunda dosis fueron del 87,9% y el 91,9% para los grupos de 3 y 6 μg, respectivamente. Además, las concentraciones de anticuerpos neutralizantes contra el SARSCoV-2 y las tasas de seroconversión se evaluaron mediante un ensayo de microneutralización con SARS-CoV-2 y una prueba de neutralización por reducción de placa usando tres cepas diferentes para el desafío viral. Las concentraciones de anticuerpos neutralizantes fueron significativamente más altos en los dos grupos de Alum-imidazoquinolina que en el grupo de la vacuna con solo Algel, mientras que las tasas de seroconversión neutralizante fueron del 93,4% y el 86,4% en los grupos de 3 y 6 μg adyuvantes con Alum-imidazoquinolina, respectivamente, frente al 86,6% en el grupo de 6 μg solo de Algel.
Un estudio clínico de fase 3, aleatorizado, doble ciego, para evaluar la eficacia, seguridad e inmunogenicidad de este candidato comenzó el 23 de octubre (número de registro: CTRI / 2020/11/028976) en India.
Un total de 25.800 participantes adultos recibirán 3 μg de la forma con adyuvante del candidato o solución salina tamponada con fosfato con adyuvante como placebo, en un régimen de estimulación inicial de dos inmunizaciones intramusculares separadas por 4 semanas. La vacuna candidata contiene 6 μg de virus inactivado por vacunación. Los resultados iniciales del ensayo clínico de Fase 3 se esperan en el primer trimestre de 2021. El 3 de enero de 2021, India otorgó la EUA a BBV152, aunque todavía se reclutan participantes para los ensayos de eficacia y seguridad de Fase 3.
Vacunas de subunidades proteicas
NVX-CoV2373 (Novavax): De manera similar a las vacunas de patógenos inactivados, los candidatos a subunidades de proteínas generalmente exhiben un perfil de seguridad extremadamente favorable, pero requieren múltiples dosis de refuerzo y provocan respuestas celulares de bajo grado.
Novavax, con sede en Maryland, ha desarrollado una nanopartícula de glicoproteína recombinante SARS-CoV-2 S de prefusión de longitud completa expresada en un sistema de baculovirus-Sf9 y se administra con un adyuvante llamado Matrix M1. El adyuvante Matrix M1 basado en saponina se utiliza precisamente para abordar la ausencia de respuestas inmunes mediadas por células que caracterizan a las vacunas de subunidades proteicas.
Novavax lanzó un ensayo de Fase 1/2 cuyos resultados se publicaron en el New England Journal of Medicine el 2 de septiembre. Un total de 131 adultos sanos fueron asignados aleatoriamente para recibir dos administraciones de la vacuna con o sin el adyuvante o un placebo. Los efectos adversos producidos fueron nulos o leves y de corta duración. La adición de adyuvante mejoró las respuestas inmunes provocadas por el candidato a vacuna y dio como resultado respuestas celulares que exhibieron un fenotipo sesgado de Th1. Los anticuerpos anti-S IgG y neutralizantes inducidos por la vacunación superaron a los detectados en sueros convalecientes de pacientes con COVID-19.
El 23 de septiembre, Novavax lanzó una prueba de Fase 3 que tiene como objetivo inscribir hasta 9000 voluntarios en el Reino Unido (Número de registro: 2020-004123-16) y planea expandirse en Estados Unidos, India y otros países. Durante el mismo mes, Novavax estableció una colaboración con el Serum Institute of India que permitirá la producción de hasta 2 mil millones de dosis al año. El 28 de diciembre, Novavax inició un ensayo de Fase 3 en los EE. UU. con el objetivo de reclutar a 30.000 participantes, la mitad de los cuales recibirán 5 μg de nanopartícula de glicoproteína recombinante SARS-CoV-2 S de prefusión completa con 50 μg de adyuvante Matrix M1 (Identificador de ensayo clínico: NCT04611802).
De la lista de las diez principales vacunas candidatas, cinco (ARNm-1273, ARNm-BNT162b2, AZD1222, JNJ-78436735, NVX-CoV2373) forman parte de Operation Warp Speed, una iniciativa que se ha fijado el objetivo de entregar 300 millones de dosis de vacunas seguras y eficientes para mediados de 2021 en los EE. UU.
ZF2001 (Biofarmacéutica Anhui Zhifei Longcom / Academia China de Ciencias Médicas): El último candidato a vacuna de subunidad que ingresó en los estudios clínicos de Fase 3 es el antígeno dimérico RBD adyuvante diseñado por Anhui Zhifei Longcom Biopharmaceutical y el Instituto de Microbiología de la Academia China de Ciencias Médicas. El estudio clínico de fase 3 se lanzó en diciembre y se llevará a cabo inicialmente en China y Uzbekistán, mientras que Indonesia, Pakistán y Ecuador seguirán como sitios de estudio (Identificador de ensayo clínico: NCT04646590 y Número de registro: ChiCTR2000040153). El diseño del estudio implica el reclutamiento de 22.000 voluntarios de China y 7.000 sujetos fuera de China para un total de 29.000 voluntarios. Todavía no hay resultados publicados sobre este candidato; sin embargo, los datos de su ensayo clínico de Fase 2 controlado con placebo (Identificador de ensayo clínico: NCT04466085) realizado en un total de 900 participantes de entre 18 y 59 años sugieren que se evalúe un régimen de 2 o 3 dosis. Cada inmunización estará separada por la siguiente por 4 semanas.
Nombre desconocido (Sanofi Pasteur / GlaxoSmithKline): Este candidato está diseñado de manera similar a la vacuna tetravalente FluBlok producida por Sanofi. Sanofi utiliza un sistema de expresión de baculovirus para transferir la información genética del inmunógeno en células de insectos lepidópteros que posteriormente expresan altos niveles del antígeno codificado, en este caso, la proteína S del SARS-CoV-2. GSK está proporcionando su AS03 (Adjuvant System 3) adyuvante a base de escualeno que se ha utilizado con éxito en varias vacunas desarrolladas por GSK, como la vacuna contra la influenza pandémica A (H1N1) llamada Pandemrix. El 3 de septiembre, Sanofi anunció que entrarían en ensayos clínicos combinados de Fase 1/2 con su vacuna candidata. Este ensayo se realizó con la participación de 440 participantes en 11 sitios de investigación en los Estados Unidos (número de registro: NCT04537208). Los participantes se dividieron en tres grupos diferentes y recibieron una o dos dosis de la vacuna, o un control de placebo, respectivamente. Sin embargo, el 11 de diciembre Sanofi y GSK anunciaron que su candidato no logró obtener respuestas inmunes fuertes en participantes mayores de 50 años y solo mostró eficacia en adultos de 18 a 49 años. Así, las dos empresas planean optimizar la concentración de antígeno administrada por su candidato para mejorar su inmunogenicidad e iniciar una Fase 2b con una nueva formulación que se comparará con una vacuna SARS-CoV-2 ya autorizada.
Vacunas de partículas similares a virus
CoVLP (Medicago): Las vacunas de partículas similares a virus tienen como objetivo combinar la eficacia de las vacunas de patógenos atenuados con el excelente perfil de seguridad que generalmente se encuentra en las vacunas de subunidades. La VLP muestra múltiples copias del antígeno diana en su superficie y tiene un tamaño que favorece el reconocimiento y la absorción posterior de las células presentadoras de antígeno, lo que promueve su fagocitosis, procesamiento y presentación eficientes por las células dendríticas e induce fuertes respuestas adaptativas.
El único candidato avanzado contra el SARS-CoV-2 que emplea esta estrategia es la vacuna diseñada por la empresa Medicago con sede en Quebec. El enfoque de Medicago es único, ya que utiliza la planta transfectada con virus Nicotiana benthamiana para expresar la forma de la subunidad trimérica de prefusión de la proteína S del SARS-CoV-2 y ensamblarla en la superficie de las VLP que se cosechan y se usan para la inmunización.
Se llevó a cabo un estudio clínico de Fase 1 después de inscribir a 180 participantes, de 18 a 55 años de edad, que fueron sometidos a un régimen de dos vacunas de cualquiera de las tres dosis (3,75 μg, 7,5 μg y 15 μg) por dosis. Además, cada una de las dosis se complementó con adyuvantes CpG 1018 o AS03 o se aplicó sin adyuvante. CpG 1018 es un ligando del receptor 9 tipo Toll desarrollado por Dynavax que induce respuestas celulares y humorales robustas, mientras que el adyuvante AS03 basado en escualeno está desarrollado y patentado por GSK y se ha utilizado en varios de los productos de la empresa. Los resultados de estos estudios se publicaron en línea como una preimpresión el 6 de noviembre e indican que la administración de CoVLP fue bien tolerada ya que generó solo eventos adversos transitorios de leves a moderados. Además, no hubo efecto dosis-dependiente sobre la inducción de anticuerpos neutralizantes y la adición de ambos adyuvantes indujo fuertes respuestas humorales y celulares. Sin embargo, cuando se administró junto con AS03, incluso la dosis más baja de 3.75 μg de la vacuna candidata fue capaz de provocar fuertes respuestas de células T y niveles de anticuerpos neutralizantes que fueron aproximadamente 10 veces más altos que los producidos en promedio en individuos convalecientes por COVID-19. Con base en estos resultados, el 19 de noviembre comenzó el reclutamiento una Fase 2/3 aleatorizada, ciega al observador y controlada con placebo (Identificador de ensayo clínico: NCT04636697). El estudio incluirá a 30,612 voluntarios adultos que recibirán 2 dosis intramusculares de 3.75 μg de vacuna CoVLP con 0.5 mL de adyuvante AS03 o un placebo con una diferencia de 3 semanas.
Conclusiones
El mundo está en medio de una pandemia de COVID-19 y toda la comunidad científica de vacunología está corriendo para encontrar una vacuna contra el SARS-CoV-2 que sea segura y eficaz. Actualmente hay más de 230 candidatos a vacunas en desarrollo, y varios de ellos ya han recibido autorizaciones de uso de emergencia en menos de un año desde el primer informe de una infección por SARS-CoV-2.
Los comités de ética están revisando sus protocolos de autorización y las empresas farmacéuticas han formado alianzas estratégicas con la institución desarrolladora de vacunas para aumentar la producción de vacunas candidatas en riesgo. Más de 150 países han entrado en la iniciativa COVAX y otras alianzas que tendrán como objetivo asegurar una distribución equitativa de una vacuna aprobada.
Varios gobiernos han realizado pagos por adelantado para asegurar una serie de dosis de las vacunas en desarrollo que ayudarán a volver a la normalidad anterior a COVID-19.
El desarrollo de múltiples candidatos a vacunas que empleen diferentes sistemas de administración probablemente resultará crucial en la lucha para poner fin a la pandemia de COVID-19. Por un lado, varias opciones de vacunación, si se aprueban, permitirán producir las dosis necesarias para la vacunación masiva en un plazo más corto. Por otro lado, es muy posible que diferentes plataformas de vacunas exhiban diferentes grados de protección contra grupos de población específicos con respuestas inmunes alteradas, como niños, mujeres embarazadas, poblaciones inmunodeprimidas debido a comorbilidades y grupos de edad inmunosenescente ≥65 años.
Varios aspectos críticos de la inmunidad al SARS-CoV-2 serán esclarecidos como resultado de campañas masivas de vacunación. La durabilidad de la inmunidad inducida por las diferentes estrategias de vacuna, así como los detalles finos de las respuestas inmunes provocadas, surgirán a medida que se vacunen poblaciones más grandes, incluidas las personas con inmunidad subóptima.
Referencias:
Kyriakidis NC, López-Cortés A, González EV, Grimaldos AB, Prado EO. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines. 2021 Feb 22;6(1):28. doi: 10.1038/s41541-021-00292-w. PMID: 33619260.
Tomado de: https://www.nature.com/articles/s41541-021-00292-w
Nota VACUNAS


La sostenibilidad como parte de cómo construimos futuro
Presentamos nuestro primer Informe de Sostenibilidad, un reflejo de cómo integramos la responsabilidad ambiental, social y de gobernanza en la…

Día de la obstetricia y la embarazada
¿Cómo se transforma la incertidumbre en una experiencia segura y asombrosa? Con una conexión única que hace de cada embarazo un viaje inolvidable.…

Los alimentos ricos en potasio mejoran la salud cardiovascular de las mujeres
De acuerdo con la Organización Mundial de la Salud (OMS), las enfermedades cardiovasculares (ECV) son la principal causa de muerte en todo el mundo y…
Viruela del mono: la OMS declara alerta máxima por el aumento de casos en el mundo
La Organización Mundial de la Salud ha declarado este sábado 23 de julio a la viruela de mono como una emergencia sanitaria internacional luego de un…