
Características y factores asociados con la infección, la hospitalización y la mortalidad por COVID-19 en diferentes razas y etnias
Clinical Infectious Diseases, 2021
https://doi.org/10.1093/cid/ciab154
RESUMEN
Antecedentes
Los datos sobre las características de los pacientes con COVID-19 desglosados por raza / etnia siguen siendo limitados. Evaluamos las características sociodemográficas y clínicas de los pacientes de todos los grupos raciales / étnicos y evaluamos sus asociaciones con los resultados de COVID-19.
Métodos
Este estudio de cohorte retrospectivo examinó a 629,953 pacientes evaluados para SARS-CoV-2 en un sistema de salud que abarca California, Oregon y Washington entre el 1ro. de marzo y el 31 de diciembre de 2020. Las características sociodemográficas y clínicas se obtuvieron de registros médicos electrónicos. Las probabilidades de infección por SARS-CoV-2, hospitalización por COVID-19 y muerte intrahospitalaria se evaluaron mediante regresión logística multivariante.
Resultados
570,298 pacientes con raza / etnia conocida fueron evaluados para SARS-CoV-2, de los cuales el 27.8% eran minorías no blancas. 54.645 personas dieron positivo, y las minorías representan el 50,1%. Los hispanos representaron el 34,3% de los pacientes con infección pero solo el 13,4% de las pruebas. Aunque en general eran más jóvenes que los pacientes blancos, los hispanos tenían tasas más altas de diabetes pero menos incidencia de otras comorbilidades. 8.536 pacientes fueron hospitalizados y 1.246 murieron, de los cuales el 56,1% y el 54,4% eran no blancos, respectivamente. Distribuciones raciales / étnicas de los resultados en todo el sistema de salud rastreadas con estadísticas a nivel estatal. Las mayores probabilidades de dar positivo en la prueba y la hospitalización se asociaron con todas las razas / etnias minoritarias. Los pacientes hispanos también exhibieron una mayor morbilidad, y la raza / etnia hispana se asoció con la mortalidad hospitalaria (OR: 1,39 [IC del 95%: 1,14-1,70]).
Conclusión
Las grandes disparidades en la atención médica fueron evidentes, especialmente entre los hispanos que dieron positivo en un índice más alto, requirieron un exceso de hospitalización y ventilación mecánica, y tuvieron mayores probabilidades de mortalidad hospitalaria a pesar de ser más jóvenes. Se necesitan intervenciones dirigidas, culturalmente sensibles y el desarrollo y distribución equitativa de vacunas para abordar el mayor riesgo de resultados de COVID-19 más deficientes entre las poblaciones minoritarias.
Introducción
La evidencia ha indicado que COVID-19 afecta de manera desproporcionada a pacientes de minorías raciales y étnicas. Si bien los informes han identificado diferentes tasas de infección, hospitalización y mortalidad entre las poblaciones minoritarias, hay información limitada sobre las características de los pacientes con COVID19 desglosadas por raza/etnia.
La prevalencia de comorbilidades y entornos sociales varían entre grupos raciales o étnicos.
Algunas de estas características, incluida la obesidad y el hacinamiento en las viviendas, son factores de riesgo potenciales para COVID-19 y la gravedad de la enfermedad. Por lo tanto, comprender cómo las características de los pacientes difieren entre razas/etnias y qué factores están asociados con los resultados de la enfermedad son críticos para la salud pública y el diseño de intervenciones basadas en la comunidad. Desafortunadamente, el detalle de tales características sigue siendo escaso y ciertos grupos raciales/étnicos, específicamente asiático-americanos, nativos, hawaiianos/isleños del Pacífico (NH/PI) y los indios americanos/nativos de Alaska (AI/AN) aún no se han caracterizados en detalle.
Además, aunque las características sociodemográficas y de salud varían en base a la distribución geográfica, las comparaciones entre varios estados son limitadas. Por tanto, el objetivo de este estudio es examinar las características y los factores asociados con la infección por SARS-CoV-2, la hospitalización con COVID-19 y mortalidad hospitalaria en una gran población diversa de pacientes en un sistema de salud basado en California, Oregon y Washington.
Método
Diseño, entorno y población del estudio
Este estudio de cohorte retrospectivo incluyó pacientes de California, Oregon y Washington que
fueron analizados para SARS-CoV-2 con el ensayo PCR, en la instalación de Providence St. Joseph Heath (PSJH) entre el 1 de marzo de 2020 y 31 de diciembre de 2020. Los resultados hospitalarios se controlaron hasta el 31 de enero de 2020. PSJH es uno de los los sistemas de salud más grandes de los EE. UU.
Los datos demográficos y clínicos de los pacientes se extrajeron del registro de salud electrónico Epic de PSJH sistema. Se consideró que los pacientes con una prueba de PCR positiva para el SARS-CoV-2 tenían una infección confirmada por el SARSCoV-2.
Los datos demográficos extraídos incluyeron edad, sexo, raza, etnia y plan de seguro. 85
los pacientes sin sexo fueron excluidos del estudio. Incluyeron condiciones médicas subyacentes que previamente se han asociado con COVID-19. El índice de comorbilidad de Charlson (ICC) se utilizó para capturar el riesgo de múltiples comorbilidades. También incluyeron la obesidad y la hipertensión, que no forman parte de la ICC. Los datos de encuentros con pacientes hospitalizados
incluyeron signos vitales, valores de laboratorios de referencia, uso de oxígeno suplementario, diagnósticos agudos (COVID-19: U07.1; infección de las vías respiratorias inferiores: J22; síndrome de dificultad respiratoria aguda: J80; insuficiencia respiratoria: J96; neumonía: J12.89), duración de la estadía, traslado a la unidad de cuidados intensivos y disposición para recibir el alta
Para comparar con los datos a nivel estatal, los casos de COVID-19 y las muertes en California, Oregon y Washington se obtuvieron de COVID Racial Data Tracker, que agrega datos históricos de
agencias estatales. Los datos sobre la hospitalización por COVID-19 para California y Oregon se obtuvieron de COVID-NET del Centro para el Control de Enfermedades, mientras que los datos de Washington se obtuvieron del COVID Radical Data Tracker.
Resultados
Características de los pacientes sometidos a pruebas de detección del SARS-CoV-2
Se incluyó un total de 629,953 pacientes evaluados para SARS-CoV-2.
570.298 pacientes (90,5%) informaron raza/etnia, de los cuales 72,2% eran blancos, 13,4% eran hispanos, 5,4% eran asiáticos, 3,8% negros, 0,9% fueron AI / AN, 0.6% fueron NH / PI y 3.7% fueron Otros. La edad media de los pacientes evaluados fue de 51,5 ± 19,4 años y el 57,0% eran mujeres. La mayoría de los pacientes (53,7%) tenían seguros (Medicaid: 30.0%; Medicare: 23.7%), con porcentajes más altos de hispanos, negros y pacientes AI / AN con Medicaid que pacientes blancos, asiáticos y NH / PI. Las comorbilidades más comunes fueron obesidad (37,1%), hipertensión (23,3%), diabetes (9,4%) y asma (6,5%).
La puntuación media en el índice de comorbilidad de Charlson (ICC) fue de 1,0 (IC del 95%: 0,0-3,0).
Características de los pacientes positivos para la infección por SARS-CoV-2
Unos 54.645 pacientes (8,7%) de los 629.953 pacientes evaluados para SARS-CoV-2 fueron positivos. Entre los cuales, 49.081 eran pacientes con raza/etnia conocida, la tasa de resultados positivos en las pruebas fue mayor para las minorías que los pacientes blancos (5,9%). Los pacientes hispanos y NH/PI tuvieron las tasas más altas (22,1% y 13,5%, respectivamente).
En consecuencia, la composición racial/étnica del SARS-CoV-2 en los pacientes infectados fue de 49,9% blancos, 34,3% hispanos, 5,0% asiáticos, 4,8% otros, 4,2% negros, 1,0%, pacientes NH/PI (1,4%) y 0,9% con IA/AN.
Entre los pacientes infectados con SARS-CoV-2, la edad media fue de 47,8 ± 19,2 años y el 52,6% eran mujeres.
En comparación con los pacientes blancos, las edades medias fueron más bajas entre los pacientes de minorías en raza/etnia, a excepción de los asiáticos, que tenían una edad media similar. Los puntajes del CCI también fueron más bajos entre los pacientes de minorías. Sin embargo, la prevalencia de diabetes fue mayor entre los pacientes de minorías.
Además, en relación con los pacientes blancos, los pacientes hispanos, negros, NH / IP y AI / AN tuvieron mayor prevalencia de la obesidad. Los pacientes asiáticos, negros, NH / IP y AI / AN tuvieron una mayor prevalencia de hipertensión; y los pacientes de raza negra, NH / PI y AI / AN tenían una mayor prevalencia tanto de asma como de enfermedad del riñón.
Los pacientes hispanos, negros, NH / PI y AI / NH tenían más probabilidades de tener seguro de Medicaid que los pacientes blancos y asiáticos. Todos los pacientes pertenecientes a minorías tenían más probabilidades que los pacientes blancos de residir en barrios con mayores porcentajes de viviendas hacinadas y minorías. Hispanos, asiáticos y pacientes negros, en particular, también tenían más probabilidades de vivir en vecindarios con un mayor porcentaje de población con dominio limitado del inglés. Todos los pacientes de minorías tenían más probabilidades de haberse realizado la prueba en el departamento de emergencias que los pacientes blancos.
Características de los pacientes hospitalizados con COVID-19
El 15,6% (n = 8.536) de los pacientes que dieron positivo por COVID-19 fueron hospitalizados. 8.210 pacientes (96,2%) tenían raza / etnia conocida, de los cuales el 45,7% eran blancos, el 35,3% eran hispanos, el 6,5% eran asiáticos, el 4,5% eran negros, el 1,2% eran NH / PI, el 0,9% eran AI / AN y el 5,9% eran “otros” (Tabla 1). La edad media de todos los pacientes fue de 64,7 ± 17,6 años y el 54,5% eran varones. La puntuación media del CCI fue de 3,0 (2,0-6,0) y las comorbilidades más frecuentes fueron obesidad (42,4%), hipertensión (40,2%) y diabetes (28,3%).
Los pacientes de raza blanca tenían la edad media más alta (70,5 ± 16,2 años; Tabla 3) y la puntuación media del CCI (4,0 [3,0- 7.0]). Los pacientes con NI / IP tenían la edad media más baja (55,9 ± 17,3 años) y la puntuación media más baja del CCI (2,0 [1.0-5.0]).
La prevalencia de obesidad fue más alta entre pacientes con AI / AN, NH / PI e hispanos y más baja entre los pacientes asiáticos. Sin embargo, los hispanos tenían la prevalencia más baja de hipertensión mientras los pacientes asiáticos y negros tuvieron la más alta. La mayoría de los pacientes hispanos, negros y AI / AN contaban con Medicaid; y los pacientes hispanos, negros y asiáticos residían en vecindarios con mayor porcentaje de viviendas hacinadas, minorías y hablantes de inglés limitados que otros grupos raciales/étnicos.
Al ingreso, un mayor porcentaje de pacientes hispanos (11,1%) que de pacientes blancos (6,7%) tuvieron una puntuación de cinco o más en la escala de progresión clínica de la OMS, y un porcentaje más alto de hispanos que de pacientes blancos tenían fiebre, baja saturación de oxígeno y alta frecuencia respiratoria.
Durante el transcurso de la hospitalización, un porcentaje más alto de pacientes hispanos (18,5%) que blancos (11,1%) necesitaron ventilación mecánica.
Características de los pacientes infectados con SARS-CoV-2 a lo largo del tiempo
A lo largo de la pandemia, las tasas de pruebas positivas, hospitalización y mortalidad hospitalaria por 100,000 pacientes fueron en promedio más altos para los pacientes negros, hispanos, NH / IP y AI / AN que los asiáticos y pacientes blancos (ver Figura 1 debajo).

Figura 1: Características de los pacientes con COVID-19 a lo largo del tiempo (promedio móvil de 7 días). La columna de la izquierda representa a los pacientes que dieron positivo en la prueba de SARS-CoV-2; la columna del medio representa a pacientes que han sido hospitalizados por COVID-19; y la columna de la derecha representa los pacientes hospitalizados por COVID-19 que sufrieron muerte intrahospitalaria. La primera fila de cada columna representa el recuento promedio de pacientes de 7 días para el evento; la segunda fila representa la media móvil de 7 días de edad de los pacientes; y la tercera fila representa la tasa media de eventos de 7 días por cada 100.000 pacientes. La velocidad por cada 100.000 pacientes se calculó sobre el total de pacientes bajo atención desde 2019 para cada raza / etnia.
Los pacientes hispanos, en particular, tuvieron las tasas más altas, especialmente durante el resurgimiento de junio-julio y noviembre-diciembre de COVID-19. La edad promedio de los pacientes infectados con SARS-CoV2 generalmente disminuyó con el tiempo, mientras que la edad promedio de los pacientes con COVID-19 hospitalizados y los pacientes que experimentaron mortalidad intrahospitalaria se mantuvieron relativamente constantes.
Comparación con casos de SARS-CoV-2 a nivel estatal, hospitalización por COVID-19 y muerte
La distribución de la raza/etnia entre los pacientes de este estudio reflejó generalmente a nivel estatal las distribuciones para los resultados de COVID-19, incluida la mortalidad en la que los datos a nivel estatal capturaron muertes intrahospitalarias y extrahospitalarias. Altas proporciones de hispanos fueron constantemente observados tanto en los datos estatales como en el sistema de salud. Las diferencias en distribuciones raciales/étnicas entre la población del sistema de salud y la población de la zona de influencia fueron consistentes o menores que las diferencias observadas a nivel estatal.
Factores asociados con la infección por SARS-CoV-2
Las poblaciones minoritarias que incluyen hispanos, negros, asiáticos, NH / PI y AI / AN tenían mayores probabilidades de infección por SARSCoV-2 en comparación con los blancos en el análisis ajustado y no ajustado. En el análisis multivariado ajustado, el aumento de la probabilidad de resultados positivos de SARS-CoV-2 se asoció de forma independiente con hispanos (OR [IC del 95%]: 3,09 [2,99-3,18]), NH / PI (2,23 [2,01-2,48]), negros (1,35 [1,28 – 1,43]), raza / etnia asiática (1,31 [1,25 – 1,38]) y otra (1,69 [1,6 – 1,78]) con pacientes blancos como categoría de referencia.
El aumento de la edad y el cuadrado de la edad, sexo masculino, sobrepeso, obesidad (todas las categorías), Seguro de Medicaid, falta de seguro y residencia en un vecindario con mayor porcentaje y los individuos con dominio limitado del inglés también se asociaron de forma independiente con mayores probabilidades de infección. Sin embargo, la puntuación CCI más alta, el seguro de Medicare y el ingreso medio más alto fueron asociado con menores probabilidades de infección por SARS-CoV-2 positiva.
Factores asociados con la hospitalización por COVID-19
Las razas / etnias minoritarias también se asociaron con mayores probabilidades de hospitalización por COVID-19. En el análisis multivariado, la razón de posibilidades para la hospitalización por COVID-19 fue más alta entre pacientes con NH / IP (2,01 [1,55 – 2,61]), seguidos de asiáticos (1,62 [1,43 – 1,84]), AI / AN (1,56 [1,17 – 2,06]), Otros pacientes (1,32 [1,16 – 1,5]), hispanos (1,31 [1,22- 1,42]) y negros (1,18 [1,02 – 1,36]).
La mayor edad, sexo masculino, seguro público, no contar con seguro, un puntaje CCI más alto, bajo peso, tener obesidad de clase 2 o 3, hipertensión y residencia en un barrio con mayores tasas de dominio limitado del inglés también se asoció de forma independiente con mayores probabilidades de hospitalización.
Factores asociados con la mortalidad hospitalaria en los ingresos por COVID-19
En el análisis multivariado ajustado, la raza/etnia hispana se asoció significativamente con un aumento en la probabilidad de mortalidad hospitalaria (1,41 [1,15 – 1,71]). Otras razas/etnias minoritarias, sin embargo, no significativamente asociados. La mortalidad hospitalaria también se asoció de forma independiente con la edad; obesidad de clase 3; seguro público; puntuación más alta en la escala de progresión clínica de la OMS y en el CCI; y recuento alto de glóbulos blancos, linfocitos bajo, recuento bajo de plaquetas, AST alto, nitrógeno ureico en sangre alto, alta creatinina y alta bilirrubina. Un análisis de interacción de raza/etnia y edad identificado más a fondo, mostró un aumento desproporcionado de las probabilidades de mortalidad hospitalaria entre los pacientes hispanos a medida que aumenta la edad (1,30 [1,04 – 1,62]; Pinteraction = 0,021)
Discusión
Este estudio examinó las características y los resultados clínicos de 629,953 pacientes evaluados para el SARSCoV-2 en California, Oregon y Washington. En general, destacaron cómo las características de los pacientes infectados por SARSCoV-2 varían según la raza/etnia y muestran asociaciones diferenciales de COVID-19 hospitalizaciones, morbilidad y mortalidad en subpoblaciones raciales/étnicas. Mientras que los pacientes de la raza/etnia minoritaria representó el 27,8% de los pacientes evaluados para el SARS-CoV-2, constituyeron el 50,1% de los pacientes infectados con SARS-CoV-2 y el 54,3% de los pacientes hospitalizados por COVID-19. Pacientes hispanos en particular representaron el 34,3% de los infectados con SARS-CoV-2 y el 35,3% de los pacientes hospitalizados a pesar de lo que representa solo el 13,4% de los pacientes evaluados, un patrón consistente con las estadísticas a nivel estatal.
La raza / etnia se asoció con la infección por SARS-CoV-2 y la hospitalización por COVID-19, con
el aumento de las probabilidades de ambos resultados entre los hispanos y los pacientes con NH / IP.
La raza/etnia también se asoció significativamente con un aumento de las probabilidades de mortalidad hospitalaria. Estos hallazgos muestran la carga desproporcionada de COVID-19 que ocurre entre los hispanos en el oeste de EE. UU, a pesar de ser más joven y tener, en general, menos comorbilidades que los pacientes blancos.
Comprender las disparidades raciales y étnicas en los casos y resultados de COVID-19 es importante para comprender la naturaleza de la enfermedad y orientar los esfuerzos de intervenciones de prevención de salud pública. Este estudio detalló y comparó exhaustivamente los aspectos clínicos y las características epidemiológicas de COVID-19 en todas las razas/etnias principales, destacando en particular asiáticos, NH/PI, y pacientes AI/AN que han estado infrarrepresentados en la literatura sobre COVID-19 hasta la fecha.
Al hacerlo, muestran que estas poblaciones, particularmente los pacientes NH/IP, tienen probabilidades similares o más altas de infección por SARS-CoV-2 y hospitalización como pacientes negros e hispanos. Al mismo tiempo, mostraron que los factores asociados con la gravedad de COVID-19 varían según la raza/etnia.
Estos hallazgos demuestran la necesidad para enfocar aún más los recursos en abordar el COVID-19 en todas las comunidades minoritarias a través de esfuerzos tales como comunicaciones de salud pública culturalmente apropiada, recopilación de datos, mejora del acceso a las pruebas y
posiblemente una intervención activa más temprana en el curso de la enfermedad.
A diferencia de estudios anteriores, en este estudio se encontró una asociación significativa entre la raza/etnia hispana y la mortalidad hospitalaria, así como una interacción significativa entre la raza/etnia hispana y la edad, lo cual significa que la relación entre la raza/etnia hispana y la mortalidad hospitalaria está moderada por la edad. Las características clínicas de los pacientes hispanos al ingreso hospitalario sugieren que éstos se presentan con una enfermedad más grave que los pacientes blancos, ya que tienen más probabilidad de tener fiebre, baja saturación de oxígeno y altas tasas de respiración. El mayor porcentaje de pacientes hispanos presentado con puntajes de la escala de progresión clínica de la OMS de cinco o más refleja una necesidad de flujo alto oxígeno suplementario o ventilación mecánica.
Si bien este estudio no puede identificar las causas detrás de las asociaciones observadas, se han sugerido ciertos determinantes sociales, estructurales o biológicos en la salud.
Los determinantes sociales y estructurales podrían incluir el riesgo de ocupación y el acceso limitado a la atención médica. Se necesitan estudios adicionales para identificar los factores causales que impulsan las disparidades en COVID-19.
Las disparidades raciales/étnicas continúan persistiendo mientras dura la pandemia. Nuestro análisis temporal reveló que los resurgimientos de COVID-19 particularmente agobiaban a los pacientes de minorías.
A medida que progresa el desarrollo de las vacunas y tratamientos para COVID-19, se ve reflejada la necesidad de contar con estudios clínicos que cuenten con una representación significativa de diversas poblaciones, particularmente de aquellos afectados desproporcionadamente por COVID-19. Dicha representación también puede ayudar a generar confianza pública en las vacuna y tratamientos. La distribución de vacunas también debe tener en cuenta la equidad racial, ya que puede ayudar a mitigar el impacto desproporcionado del virus.
Desafortunadamente, las poblaciones minoritarias enfrentan varias barreras para la vacunación, que incluyen falta de acceso a la asistencia sanitaria, desconfianza en el sistema sanitario, barreras de comunicación y desinformación. Por lo tanto, deben destinarse recursos adicionales a llegar a las poblaciones de alto riesgo, generar confianza y reducir las barreras a la vacunación entre las minorías.
En conclusión, estos resultados destacan cómo el impacto de COVID-19 varía según la raza/etnia en una gran área geográfica.
Referencia:
Characteristics and Factors Associated with COVID-19 Infection, Hospitalization, and Mortality Across Race and Ethnicity, Clinical Infectious Diseases, 2021
Chengzhen L Dai, M.S, Sergey A Kornilov, Ph.D, Ryan T Roper, M.S, Hannah Cohen-Cline, Ph.D, Kathleen Jade, N.D, Brett Smith, M.S, James R Heath, Ph.D, George Diaz, M.D, Jason D Goldman, M.D., M.P.H, Andrew T Magis, Ph.D, Jennifer J Hadlock, M.D,
Características y factores COVID-19

Importancia de las intervenciones no farmacéuticas para disminuir el inóculo viral y reducir la susceptibilidad a la infección por SARS-CoV-2 y la gravedad de la enfermedad
Dada la heterogeneidad tanto en la gravedad de la enfermedad como en la incidencia de COVID-19 en todo el mundo, algunos expertos han sugerido que la adhesión a las intervenciones no farmacéuticas (p. ej., distanciamiento social y uso de mascarillas) es un factor importante que contribuye a estas diferencias observadas. Las intervenciones se han infrautilizado gravemente como parte de la respuesta al COVID-19 en los EE. UU., especialmente con los aumentos posteriores de la infección de la enfermedad. Un promedio del 49% de los estadounidenses informó haber usado máscaras faciales diariamente durante los meses de junio a agosto de 2020, en comparación con el 95% observado en Hong Kong y el 100% reportado en Vietnam en el mismo período. Una hipótesis emergente es que el inóculo viral y las intervenciones que podrían disminuirlo podrían no solo limitar las infecciones, sino también conducir a una enfermedad COVID-19 menos grave si estas intervenciones no logran prevenir la infección.
En el artículo publicado por Spinelli MA y colaboradores en febrero del 2021 que se resume a lo largo de esta nota, se revisan los datos que respaldan la importancia del inóculo viral para la susceptibilidad a infecciones virales respiratorias, gastrointestinales y de transmisión sexual, y la evidencia disponible que vincula el inóculo con la gravedad de la enfermedad. También se argumenta que, incluso mientras se implementan vacunas seguras y efectivas, las intervenciones no farmacéuticas continuarán desempeñando un papel esencial y continuo en la supresión de la transmisión del SARS-CoV-2 y cualquier otra mutación.
La importancia del inóculo de patógenos (es decir, el número de organismos a los que está expuesto un huésped en función de la concentración, duración y carga viral del material infeccioso de la fuente) sobre la probabilidad resultante de infección se ha descrito bien en humanos durante varios años para varios patógenos virales, como los virus de la influenza, virus sincitial respiratorio, adenovirus, enterovirus, poliovirus, rinovirus y rotavirus, y también para varias bacterias y parásitos, particularmente en el contexto de los alimentos.
Dada la gravedad de la enfermedad asociada con la infección por SARS-CoV-2, los estudios de provocación en humanos son controvertidos. Sin embargo, en un modelo animal de infección por SARS-CoV-2, los hámsteres sirios se infectaron con éxito con dos dosis diferentes de SARS-CoV-2, intranasal e intraocularmente, y la dosis más alta se asoció con una mayor pérdida de peso y anomalías pulmonares más graves en las imágenes de tórax. En otro experimento, cuando se colocó una máscara quirúrgica que separaba las jaulas de hámsteres sirios infectados y no infectados, solo seis (25 %) de 24 hámsteres del grupo sano, estaban infectados, en comparación con diez (67%) de los 15 animales de control a los que no se le agregó la máscara entre ambos grupos (p = 0,018).
Una especie de experimento natural ocurrió en los Alpes suizos entre el 25 de marzo de 2020 y el 14 de abril de 2020, en dos cohortes de soldados homogéneas espacialmente separadas de edad similar (edad media 21 años) y sin comorbilidades sustanciales. Después de un brote de COVID-19 ocurrido en uno de los grupos, en ambos se implementó el distanciamiento físico y el uso de mascarilla quirúrgica. Un brote de COVID-19 en el grupo previamente no afectado ocurrió después de la implementación de esta política, con 13 (15%) de 88 soldados asintomáticos que luego se confirmó que tenían COVID-19 mediante pruebas masivas (66 [43%] de 154 no fueron evaluados) y ninguno de los 154 reclutas desarrolló síntomas. En la cohorte impactada antes de que se implementara el uso de la máscara y el distanciamiento social, 102 (47%) de los 215 soldados que dieron positivo en la prueba eran sintomáticos (132 [37%] de 354 no fueron evaluados).
Otro estudio que reclutó a participantes con infección por SARS-CoV-2 confirmada por PCR y sus contactos cercanos durante el brote de SARS-CoV-2 en la primavera de 2020 en Cataluña, España, realizó un análisis post-hoc de la dinámica de transmisión en un ensayo aleatorizado por grupos en la prevención posterior a la exposición con hidroxicloroquina. El equipo visitó casos y contactos en hogares u hogares de ancianos desde el 17 de marzo de 2020 hasta el 28 de abril de 2020, y midió las cargas virales del SARS-CoV-2 de hisopos nasofaríngeos en los días 1 y 14. Este estudio encontró una relación dosis-respuesta entre la carga viral del caso índice (primer caso) y la probabilidad de enfermedad sintomática entre los contactos. La carga viral del caso índice se relacionó proporcionalmente con la transmisibilidad e inversamente con la duración del período de incubación por el que pasó el contacto infectado, con cargas virales índice más altas en los casos asociados a períodos de incubación más cortos entre los contactos. Finalmente, los datos epidemiológicos muestran un mayor número de reproducción básica (R0) del SARS-CoV-2 (número promedio de casos nuevos que genera un caso dado, a lo largo de un período infeccioso) en comparación con el coronavirus del síndrome respiratorio de Oriente Medio o el virus del síndrome respiratorio agudo severo.
Intervenciones no farmacéuticas para reducir el inóculo viral
Mascaras: Las mascarillas quirúrgicas que usan las personas infectadas reducen la transmisión al bloquear la liberación de viriones en el aire, como se ha demostrado para los coronavirus o los virus de la influenza. Ya se disponía de pruebas sobre la capacidad de las cubiertas faciales de tela para reducir también el tamaño del inóculo viral interno para otros virus respiratorios, así como para las gotas y aerosoles que simulan el SARS-CoV-2. La creciente evidencia de la investigación en ciencias físicas sobre cómo las máscaras de tela pueden proteger al usuario, así como la evidencia de larga data sobre cómo las máscaras protegen a otros, llevó a un cambio en la guía de los Centros de EE. UU. para el Control y la Prevención de Enfermedades. Así fue como el 20 de noviembre de 2020, la guía indicó que las máscaras protegen tanto al usuario como a los demás, lo que podría ayudar a aumentar el cumplimiento del uso de la máscara en los EE. UU.
En lugar de criticar la eficacia de las máscaras de tela, se deben invertir en la producción de cubiertas quirúrgicas o de otro tipo de alta calidad para aumentar su disponibilidad fuera de los entornos de atención médica. Estandarización de las recomendaciones para las mascarillas quirúrgicas (que utilizan filtración electrostática) y, si no están disponibles, máscaras de tela de alta calidad (al menos de dos capas y de alto número de hilos), reducirá la confusión. La disponibilidad y la provisión uniforme de cubiertas faciales efectivas y producidas de manera consistente también podrían reducir algunas dudas sobre el uso de mascarillas porque una mayor eficacia percibida podría aumentar el cumplimiento en poblaciones con menos adhesión al uso de las mismas.
Un estudio de modelado realizado en los EE. UU. ha encontrado una correlación entre el uso de mascarillas universales y una menor necesidad de encierros y pérdidas económicas asociadas.
Distanciamiento y ventilación: En general, se ha demostrado que el SARS-CoV-2 tiene concentraciones de ARN más altas, o un inóculo viral más alto, a distancias más cercanas a una fuente infectada o más cerca de las áreas de atención al paciente de COVID-19. Un estudio de muestreo de aire dentro de un hospital de EE. UU. en las habitaciones de pacientes con COVID-19 mostró concentraciones de ARN más altas con muestreadores de aire personales en comparación con muestreadores de aire del dormitorio o en los pasillos. En otro estudio, en un hospital en Wuhan, China, dos (18%) de 11 muestras de aire recolectadas cerca de pacientes con una infección por COVID-19 en la sala general tenían ARN detectable, en comparación con ninguna de las cinco muestras recolectadas 2,5 m de distancia de los pacientes. En la unidad de cuidados intensivos del mismo hospital, ocho (44%) de las 18 muestras recogidas a 2,5 m del paciente fueron positivas, mientras que sólo una (13%) de las ocho muestras recogidas a 4,0 m fueron positivas.
La ventilación para reducir la exposición a partículas virales ha sido bien descrita para virus respiratorios. Alentar a que las interacciones humanas sucedan principalmente en espacios al aire libre y proporcionar cambios estructurales y de ingeniería para aumentar la ventilación en los espacios interiores son intervenciones no farmacéuticas importantes.
Es importante señalar que la eficacia de las intervenciones no farmacéuticas aumentará cuando se combinen múltiples estrategias, sin que ninguna estrategia única confiera una eficacia del 100% en la prevención de la transmisión del SARS-CoV-2.
Intervenciones no farmacéuticas y efectividad de la vacuna
La eficacia de una vacuna contra el SARS-CoV-2 podría verse potencialmente afectada por la carga de la enfermedad COVID-19 a nivel poblacional. La eficacia indirecta de la vacuna (eficacia de la vacuna poblacional) ocurre cuando una vacuna previene la enfermedad en aquellos que no están vacunados a través de la suficiente inmunidad poblacional. La continuación de las intervenciones no farmacéuticas será particularmente importante para los grupos susceptibles que no desarrollan una respuesta inmune fuerte a una vacuna contra el coronavirus y para aquellos que rechazan una vacuna. La propagación incontrolada del SARS-CoV-2 en gran parte de los EE. UU. podría limitar la eficacia inicial de una vacuna contra el SARS-CoV-2.
Las noticias recientes sobre la alta eficacia de las vacunas de ARNm Moderna y Pfizer / BioNTech para el SARS-CoV-2, así como de las vacunas AstraZeneca, Novavax, Johnson y Johnson y Sputnik V, son esperanzadoras y emocionantes. Sin embargo, los criterios de valoración de los ensayos de todas estas vacunas fueron principalmente, la prevención de la enfermedad sintomática (en la que cada una de las vacunas de ARNm mostró una eficacia superior al 94% frente a un placebo). Dado que no se puede descartar una infección asintomática en los pacientes que recibieron la vacuna, será necesario mantener la adhesión continua a las intervenciones no farmacéuticas (incluso por parte de los vacunados) hasta que se controle la pandemia y se logre la vacunación generalizada. A su vez, debido a que los grupos de menor prioridad, como los jóvenes, los sanos y las personas que no trabajan en los servicios esenciales, pueden sufrir retrasos en la oferta de vacunas, las intervenciones no farmacéuticas seguirán siendo fundamentales en el futuro próximo.
Mientras se construye la infraestructura para almacenar y administrar una vacuna a gran escala, se deben realizar simultáneamente inversiones en el estudio científico, la producción y la promoción de intervenciones no farmacéuticas, como máscaras estandarizadas, para prevenir la transmisión continua del SARS-CoV-2.
En base a todo lo mencionado, las intervenciones no farmacéuticas, incluido el distanciamiento social, el uso de mascarillas y la mejora de la ventilación, especialmente si se asocian con un mayor cumplimiento en entornos con transmisión no mitigada de SARS-CoV-2, podrían marcar una diferencia importante y positiva en la gravedad y transmisibilidad del COVID-19 en todo el mundo.
Referencia:
Spinelli MA, Glidden DV, Gennatas ED, Bielecki M, Beyrer C, Rutherford G, Chambers H, Goosby E, Gandhi M. Importance of non-pharmaceutical interventions in lowering the viral inoculum to reduce susceptibility to infection by SARS-CoV-2 and potentially disease severity. Lancet Infect Dis. 2021 Feb 22:S1473-3099(20)30982-8. doi: 10.1016/S1473-3099(20)30982-8. Epub ahead of print. PMID: 33631099; PMCID: PMC7906703.
Tomado de:
https://www.thelancet.com/journals/laninf/article/PIIS1473-3099(20)30982-8/fulltext
Importancia de las intervenciones no farmacéuticas


Tecnología Informática de Megalabs certificada ISO/IEC 20000-1:2018
El Area de Infraestructura de Tecnología Informática ha logrado la certificación ISO/IEC 20000-1:2018, emitida por LSQA, Uruguay. Es un orgullo para la compañía dado que demuestra el compromiso de la organización en la adopción de buenas prácticas que garanticen servicios de TI más efectivos, así como incorporar las mejores prácticas internacionales en la gestión de los mismos.
La norma ISO 20000-1 es un estándar generado en conjunto por la Organización Internacional de Estandarización (ISO) y por la Comisión Electrotécnica Internacional (IEC), y se utiliza para la gestión y supervisión de los servicios relacionados con las Tecnologías de la Información. Es aceptada como un referente en este campo y es reconocida a nivel mundial.
¡Felicitaciones a todo el equipo de Infraestructura de TI Corporativo!


Megalabs Colombia
Es muy grato anunciarles que Megalabs Colombia ha logrado ser calificado como Operador Económico Autorizado (OEA) para los tipos de usuario importador y exportador en la categoría de seguridad y facilitación.
Esta certificación emitida por la Dirección de Impuestos y Aduanas Nacionales (DIAN) es la máxima calidad de confianza que genera una empresa de comercio exterior ante el Estado colombiano y sus autoridades aduaneras que tiene como pilares esenciales un comercio ágil, transparente y seguro.
Es un orgullo para la compañía dado que demuestra el compromiso de la organización en la adopción de buenas prácticas que garanticen los niveles óptimos de seguridad y el mejoramiento de los flujos de información y operación de la cadena de suministro internacional, beneficiando así a todos nuestros asociados de negocio.
¡FELICITACIONES A TODO EL EQUIPO DE MEGALABS COLOMBIA!

Anticoagulación profiláctica para pacientes hospitalizados con COVID-19
Beverley J Hunt, Erich V De Paula, Claire McLintock, Mert Dumantepe
BMJ 2021;372:n487
Si bien la mayoría de las personas con COVID-19 transitan la enfermedad de forma leve, una minoría desarrolla neumonía y requiere hospitalización para tratar la hipoxia. Los pacientes hospitalizados por COVID-19 presentan un estado protrombótico con altas tasas de tromboembolismo venoso.
Existe una necesidad de contar con información consistente de expertos sobre la profilaxis antitrombótica en pacientes hospitalizados por COVID-19 para tomar acciones. De acuerdo a los factores de riesgo que califican para la profilaxis trombótica: todos los pacientes hospitalizados con neumonía por COVID-19, o que ingresan a cuidados intensivos deben recibir tromboprofilaxis.
Ensayos controlados aleatorios mostraron que la tromboprofilaxis basada en medicamentos con heparina de bajo peso molecular (HBPM) reduce el riesgo de tromboembolismo venoso en aproximadamente un 50% en pacientes hospitalizados críticos. Sin embargo, al inicio de la pandemia muchos países no realizaban profilaxis trombótica en pacientes hospitalizados por COVID-19, inclusive en aquellos casos críticos. Un pequeño estudio observacional temprano mostró que la heparina mejoró la mortalidad en pacientes críticamente enfermos con COVID-19, y este hallazgo llevó a muchas unidades de cuidados intensivos a comenzar a usar tromboprofilaxis basada en HBPM y/o aumentar la dosis de a un nivel intermedio o incluso terapéutico.
Un estudio retrospectivo (Rentsch y colegas. (doi: 10.1136 / bmj.n311)) confirma que la tromboprofilaxis se asocia con una mejoría en la mortalidad en los pacientes hospitalizados por COVID-19.
Al mismo tiempo un análisis intermedio de datos combinados de tres ensayos controlados aleatorios separados (REMAP-CAP, ATTACC y ACTIV4a) informó recientemente el impacto de diferentes dosis de anticoagulación en los resultados. Se comparó la tromboprofilaxis con heparina en dosis terapéuticas (ya sea HBPM o heparina no fraccionada) con la atención estándar local en pacientes ingresados en el hospital con COVID-19 grave o moderado. La heparina fue administrada durante 14 días, o hasta el alta hospitalaria, o después de suspender el oxígeno suplementario, según lo que ocurriera primero. Los ensayos fueron detenidos para los pacientes con COVID-19 grave porque quienes recibieron dosis terapéuticas de heparina mostraron un aumento de la mortalidad en relación con los controles y una mayor necesidad de oxígeno. El riesgo de hemorragia mayor también aumentó con respecto al grupo control.
Por el contrario, también se pausaron los estudios que reclutaban pacientes con enfermedad moderada, pero esta vez debido a una aparente superioridad de la anticoagulación a dosis terapéuticas. Los resultados se analizaron según si los niveles de dímero D eran bajos o altos en el momento de la presentación, pero los resultados fueron similares entre pacientes recibiendo profilaxis antitrombótica y aquellos que no. Esto sugirió que las pruebas de dímero D no tienen valor para evaluar el riesgo trombótico en pacientes ingresados en el hospital con neumonía COVID-19. Sin embargo, los pacientes con enfermedad moderada que recibieron tromboprofilaxis en dosis terapéuticas más fueron significativamente probable que los controles de lograr el resultado primario de supervivencia o necesidad reducida de soporte de órganos.
Si estos hallazgos se confirman, futuras investigaciones deberían considerar:
• Si el beneficio de la dosis terapéutica de HBPM o de heparina no fraccionada se limita a pacientes con COVID-19 moderado
• Si el beneficio aparente de la heparina puede estar relacionado con sus efectos antiinflamatorios y antivirales y no solo con su efecto anticoagulante
• Si la heparina influye en la tasa de inmunotrombosis
• Evaluar los efectos relativos de la tromboprofilaxis estándar versus la intermedia en el COVID-19 grave
• Evaluar los efectos relativos de la tromboprofilaxis intermedia versus la terapéutica en el COVID-19 moderado
Datos retrospectivos de tromboprofilaxis prolongada (posterior al alta de COVID-19) con HBPM o anticoagulantes orales han mostrado bajas tasas de tromboembolismo venoso. Esto requiere ensayos aleatorios que evalúen formalmente la necesidad de realizar tromboprofilaxis prolongada.
Referencia: Prophylactic anticoagulation for patients in hospital with covid-19. BMJ (Clinical Research ed.). 2021 Feb;372:n538. DOI: 10.1136/bmj.n538.
Publicación original:
https://www.bmj.com/content/372/bmj.n487
anticoagulación profilactica en covid19

Mayormente peor, ocasionalmente mejor: el impacto de la pandemia de COVID-19 en la salud mental de niños y adolescentes canadienses
European Child & Adolescent Psychiatry (2021)
https://link.springer.com/article/10.1007/s00787-021-01744-3
Resumen
Este estudio examinó el impacto de las medidas de emergencia de COVID-19 en la salud mental de niños y adolescentes para aquellos con y sin diagnósticos psiquiátricos preexistentes.
Utilizando medidas adaptadas del cuestionario CRISIS, los padres de niños de 6 a 18 años (N = 1013; 56% hombres; 62% con diagnóstico psiquiátrico preexistente) y los niños/adolescentes de 10 a 18 años (N = 385) informaron cambios en la salud mental en seis dominios: depresión, ansiedad, irritabilidad, atención, hiperactividad y obsesión/compulsión. Se calcularon los cambios en la ansiedad, la irritabilidad y la hiperactividad para los niños de 2 a 5 años utilizando el Cuestionario de Fortalezas y Dificultades. La exposición a COVID-19, el cumplimiento de las medidas de emergencia, las preocupaciones económicas por COVID-19 y el estrés por aislamiento social se midieron con el cuestionario CRISIS. Se calculó la prevalencia del cambio en el estado de salud mental para cada dominio y se utilizó una regresión logística multinomial para determinar las variables asociadas con el cambio del estado de salud mental en uno.
Dependiendo del grupo etario, entre el 67% y el 70% de los niños/adolescentes experimentaron deterioro en al menos un dominio de salud mental; sin embargo, del 19 al 31% de los niños/adolescentes experimentaron una mejoría en al menos un dominio. Los niños/adolescentes con y sin diagnósticos psiquiátricos tendieron a experimentar un deterioro durante la primera ola de COVID-19. Las tasas de deterioro fueron más altas en aquellos con un diagnóstico previo. La tasa de deterioro fue variable entre los diferentes grupos etarios y los grupos de diagnóstico psiquiátrico preexistentes: depresión 37-56%, ansiedad 31-50%, irritabilidad 40-66%, atención 40-56%, hiperactividad 23-56%, obsesiones/compulsiones 13-30%. Un mayor estrés por aislamiento social se asoció con el deterioro en todos los dominios de la salud mental (todos los OR se encontraban en un rango de 11.12–55.24). El impacto del diagnóstico psiquiátrico preexistente fue heterogéneo, asociado con el deterioro de la depresión, irritabilidad, hiperactividad, obsesión/compulsión en algunos niños (OR 1,96-2,23), pero también con una mejoría en la depresión, ansiedad e irritabilidad para otros niños (OR 2,13 –3,12). Las preocupaciones económicas se asociaron con una mejora en la ansiedad, la atención y las obsesiones/compulsiones (OR 3,97–5,57). Los niños/adolescentes con y sin diagnósticos psiquiátricos preexistentes mostraron deterioro. El deterioro se asoció con un mayor estrés debido al aislamiento social. Mejorar las interacciones sociales para niños/adolescentes será una importante estrategia de mitigación para las olas de COVID-19 actuales y futuras.
INTRODUCCIÓN
Las medidas de emergencia de COVID-19, que llevaron al cierre de escuelas, centros de recreación y actividades organizadas, han provocado una interrupción generalizada de todos los aspectos de la vida y las rutinas de los niños y adolescentes. Las medidas de emergencia de COVID-19 comenzaron en Ontario, Canadá el 12 de marzo de 2020 y permanecieron en gran parte bajo confinamiento, incluido el cierre de todas las escuelas, casi todos los centros de cuidado infantil, todas las tiendas minoristas no esenciales, muchos parques locales y provinciales, y en visitas médicas y dentales presenciales (excepto para los servicios de emergencia), hasta el 22 de junio de 2020.
Las rutinas diarias proporcionadas por la escuela y las actividades extracurriculares son fundamentales para mantener la actividad física, regular los ciclos del sueño y proporcionar interacciones sociales, todos factores clave de protección para la salud mental de niños y adolescentes. La literatura emergente de países que experimentan una alta prevalencia de COVID-19, como China, sugiere un impacto psicológico significativo en niños y adolescentes. Las preocupaciones acerca de estar infectado y vivir en un área con mayor prevalencia viral se asociaron con un aumento de los síntomas depresivos y de ansiedad entre los adolescentes (44% y 37%, respectivamente).
Los estudios publicados, sin embargo, han proporcionado poca o ninguna información sobre los impactos relativos de la exposición al COVID-19 y la implementación de medidas de emergencia, por ejemplo, el cumplimiento de las directivas y el estrés del aislamiento social en la salud mental de niños y adolescentes. Además, la exposición al virus y las medidas de emergencia impuestas pueden variar de manera diferente entre los grupos de edad (desde la infancia hasta los 18 años) y el diagnóstico previo de trastornos de salud mental, del neurodesarrollo o ambos. A su vez, mientras que la mayoría de los estudios anteriores se han centrado en la depresión y la ansiedad, este estudio examinó seis dominios de salud mental.
Varios comentarios han destacado los probables efectos nocivos de las medidas de emergencia del COVID-19 sobre la salud mental de niños y adolescentes, pero también es posible que algunos de ellos experimenten mejoras en la salud mental en comparación con su situación antes de la pandemia, debido a la reducción del estrés social y/o escolar. Comprender los factores asociados con la mejora, así como con el deterioro de la salud mental es importante para comprender la capacidad de recuperación y considerar intervenciones para mejorar los resultados de quienes experimentan deterioro de la salud mental.
El objetivo principal de este estudio fue examinar el impacto de las medidas de emergencia en el estado de salud mental en comparación con la salud mental antes de la pandemia en seis dominios: depresión, ansiedad, irritabilidad, atención, hiperactividad y obsesiones/compulsiones en niños de 2 a 18 años (según lo informado por sus padres) y en niños y adolescentes de 10 a 18 años (según lo informado por ellos mismos). Los objetivos secundarios incluyeron examinar estos impactos según cada dominio de salud mental, entre niños con y sin diagnósticos psiquiátricos y de neurodesarrollo pre-COVID, e identificar factores de riesgo y de protección para cambios en la salud mental.
MATERIALES Y MÉTODOS
Participantes:
Este estudio se incluyó en cuatro cohortes de estudio en curso: dos cohortes de salud mental y desarrollo neurológico con referencias clínicas y dos cohortes de la comunidad:
(i) SickKids Psychiatry: niños/adolescentes de 6 a 18 años en el área metropolitana de To-ronto referidos a una clínica de salud mental para pacientes ambulatorios para la evalua-ción de problemas de salud mental que incluyen, entre otros, depresión y trastornos de ansiedad, trastorno por déficit atencional/hiperactividad (TDAH), trastorno obsesivo compulsivo (TOC), trastornos de conducta disruptiva;
(ii) la red de trastornos del neurodesarrollo de la provincia de Ontario (POND): niños y adolescentes de 6 a 18 años de toda la provincia de Ontario que reciben atención en clí-nicas ambulatorias con trastornos del neurodesarrollo, incluidos los trastornos del espec-tro autista (TEA), TDAH, TOC y discapacidad intelectual;
(iii) El Grupo de Investigación Aplicada para Niños (TARGet Kids!): niños sanos recluta-dos desde el nacimiento hasta los 5 años en el área metropolitana de Toronto y que parti-cipan en una red de investigación basada en la práctica de atención primaria; y
(iv) Spit for Science: una muestra poblacional de niños/adolescentes de 6 a 18 años reclu-tados en un museo de ciencia urbana. La muestra está compuesta principalmente por ni-ños y adolescentes de la región sur de Ontario.
Estas cuatro cohortes se seleccionaron como cohortes establecidas con una base de participantes diversa y existente para este estudio sobre los efectos de COVID-19 en la salud infantil y salud mental. Incluyen niños de todos los rangos de edad hasta los 18 años. Mediante la inclusión de muestras clínicas y comunitarias (por ejemplo, niños y adolescentes con diagnósticos de salud mental y/o desarrollo neurológico anteriores a COVID), los impactos de la pandemia podrían examinarse en múltiples poblaciones importantes. A los padres que previamente habían dado su consentimiento para ser contactados, se les envió un correo electrónico sobre la participación con un enlace separado para enviar a su hijo si su hijo tenía entre 10 y 18 años y estaba interesado en participar. Este estudio fue aprobado en todas las juntas de ética de investigación institucional y todos los participantes proporcionaron consentimiento informado.
Los padres de niños de 2 a 18 años (n = 1013; tasa de respuesta del 53,2%) completaron cuestionarios en línea utilizando REDcap. Los jóvenes de 10 a 18 años (n = 347) completaron cuestionarios a través de un enlace de correo electrónico separado a la dirección de correo electrónico de sus padres. Esta muestra es una muestra de conveniencia y los participantes no fueron seleccionados para ser incluidos en función de las características específicas. Las medidas de emergencia se implementaron en Canadá a mediados de marzo de 2020. Toda la recopilación de datos mediante cuestionarios en línea se completó desde el 15 de abril de 2020 hasta el 19 de junio de 2020. Algunos datos para usar en el cálculo de la puntuación de cambio del estado de salud mental (como se describe a continuación) para niños de 2 a 5 años se recopilaron desde el 1 de agosto de 2018 hasta el 29 de febrero de 2020. El análisis se completó hasta el 31 de julio de 2020.
MEDIDAS:
Cambio de estado en la salud mental de niños/adolescentes
Entre los niños/adolescentes de 6 a 18 años, el cambio en la salud mental se evaluó por separado (por dominio) utilizando una versión adaptada de un solo ítem del Cuestionario internacional CRISIS, que mide el impacto de la pandemia para que pueda compararse entre culturas. Se preguntó a los padres y a los jóvenes: «En comparación con el tiempo ANTES de la crisis del COVID-19, ¿cómo es el estado de ánimo general de su hijo?», Utilizando una escala Likert de 5 puntos (1 = mucho peor; 5 = mucho mejor). Esto se repitió para cada uno de los 6 dominios de salud mental (depresión, ansiedad, irritabilidad, atención, hiperactividad, obsesiones/compulsiones). Investigaciones anteriores han demostrado la validez de medidas de elementos individuales similares en la evaluación del sufrimiento psicológico.
Los padres de niños de 2 a 5 años completaron el Cuestionario de fortalezas y dificultades preescolares (SDQ preescolar, edades de 2 a 4) o el SDQ (para niños de 5 años). Se utilizaron tres subescalas del SDQ / SDQ preescolar que se alinean con los dominios evaluados en los niños mayores: subescala de problemas emocionales por ansiedad, subescala de problemas de conducta por irritabilidad y subescala de hiperactividad para la hiperactividad. Los ítems de la subescala de problemas de conducta en el SDQ y SDQ preescolar se alinean más estrechamente con la irritabilidad, un síntoma central del trastorno negativista desafiante que es la presentación más común de problemas de conducta en el grupo de edad preescolar.
Demografía
Se registró el ingreso familiar informado por los padres, edad del niño, raza/etnia, sexo, identidad de género y diagnósticos psiquiátricos pre-COVID, utilizando elementos adaptados del cuestionario CRISIS, un instrumento diseñado por una colaboración internacional para examinar la salud mental durante la pandemia COVID-19, y el CRISIS-AFAR, adaptado para el autismo y las condiciones del neurodesarrollo relacionadas.
Impacto económico del COVID-19
Fue determinado utilizando dos elementos informados por los padres del cuestionario CRISIS.
Cumplimiento de las medidas de emergencia / directivas de permanencia en el hogar
Fue determinado utilizando los ítems adaptados del cuestionario CRISIS.
Exposición al COVID-19
Fue determinado por las respuestas a dos ítems adaptados del cuestionario CRISIS sobre el diagnóstico de COVID-19 en el padre, hijo o familiar. Una respuesta afirmativa a cualquiera de los elementos se codificó como «exposición».
Estrés por aislamiento social
Fue evaluado utilizando cuatro ítems del cuestionario CRISIS. Las respuestas a los ítems se proporcionaron en una escala Likert de cinco puntos (1 = nada; 5 = extremadamente) a partir de la cual se calculó la puntuación media de estrés por aislamiento social, con puntuaciones más altas indicando un mayor nivel de estrés.
Análisis de datos:
Los datos se analizaron en R y R studio versión 1.2.1335. Se utilizó mice para la imputación (n = 10) de datos de predictores y covariables a nivel de ítem faltantes, donde los participantes tenían <50% de datos faltantes en todos los ítems requeridos para el análisis. Para los predictores de escala (impacto económico de COVID-19, cumplimiento de las directivas de quedarse en casa, estrés por aislamiento social), las respuestas a los ítems se promediaron y estandarizaron en una escala de 0 a 1 para facilitar las comparaciones.
Cambio de estado de salud mental
Para las respuestas de los padres de niños de 6 a 18 años, se crearon 3 categorías usando los seis ítems sobre el cambio del estado de salud mental al combinar puntuaciones de 1 y 2 (mucho y un poco peor) en una categoría de «deteriorado»; puntuaciones de 3 (aproximadamente lo mismo) en «sin cambios»; 4 y 5 (un poco y mucho mejor) en «mejorado». Las respuestas del informe de los jóvenes se codificaron en un proceso idéntico. Para los niños de 2 a 5 años, se calculó una puntuación de diferencia utilizando las puntuaciones obtenidas durante el período de estudio y los datos recopilados en el SDQ en los 18 meses anteriores al COVID-19, de la cohorte “TARGet Kids!”. Mejorado, deteriorado y sin cambios se puso en práctica utilizando un cambio de 2 puntos como diferencia mínima clínicamente significativa en las puntuaciones del SDQ (la diferencia de ≤ – 2 indicó mejoría, la diferencia ≥ 2 indicó deterioro, y la diferencia <2 y> – 2 indicó sin cambios).
Para los niños/adolescentes de 10 a 18 años, se examinó la concordancia entre el niño y los padres informantes sobre el cambio del estado de salud mental utilizando la estadística Χ2.
La mejora o el deterioro en cualquier dominio en todos los dominios para niños/adolescentes de 6 a 18 años y para niños de 2 a 5 años se examinó con estimaciones de prevalencia. La prevalencia de calificaciones «mejoradas», «sin cambios» o «deterioradas» en los seis dominios de salud mental por informante, por grupo de edad del niño, así como en todos los participantes, y por separado en niños con y sin diagnósticos psiquiátricos pre-COVID, luego se examinaron con estimaciones de prevalencia. Diagnósticos psiquiátricos preexistentes, evaluados en el cuestionario CRISIS, se agruparon en cuatro categorías: (1) salud mental (HM: depresión, ansiedad, TOC, TDAH o trastorno del aprendizaje); (2) trastorno del espectro autista (TEA: trastorno del espectro autista, retraso en el desarrollo, otro trastorno del desarrollo neurológico); (3) trastorno comórbido de salud mental y del espectro autista/retraso en el desarrollo (MH + TEA: incluidos los diagnósticos de MH y TEA enumerados); y (4) ningún diagnóstico psiquiátrico (NO DX: participantes sin diagnóstico de MH o TEA).
RESULTADOS
Características de los participantes
La muestra incluyó a 1013 padres de niños/adolescentes de los 1905 padres que inicialmente confirmaron la recepción del correo electrónico del estudio. Los niños y adolescentes tenían entre 2 y 18 años. El 56,1% (n = 568) eran hombres. De los 763 niños y adolescentes que dieron su consentimiento para participar, 347 completaron las medidas de resultado. La edad media (DE) de niños y adolescentes fue 13,05 (2,53) y el 51,8% eran varones.
Cambio de estado de salud mental
Al examinar la prevalencia combinada en todos los dominios del deterioro del informe de los padres en cualquiera de los seis dominios evaluados, el 70,2% (651 de 927) de los niños y adolescentes de 6 a 18 años estaban peor en al menos un dominio. Entre los niños de 2 a 5 años, el 66,7% (36 de 54) estaba peor en al menos uno de los tres dominios evaluados. Para la prevalencia combinada de mejora en todos los dominios en cualquiera de los seis dominios, el 19,5% (181 de 927) de los niños y adolescentes de 6 a 18 años obtuvieron mejores resultados en al menos un dominio. Entre los niños de 2 a 5 años, al 31,5% (17 de 54) les iba mejor en al menos uno de los tres dominios.
Cambios en el estado de salud mental en todas las edades y grupos de informantes
El informe de los padres fue coherente con el autoinforme de los jóvenes sobre los cambios en la depresión para las categorías mejorías, sin cambios, deteriorado (Χ² (1) = 43,91, p <0,001). En todos los demás dominios, los tamaños de celda para «mejorías» fueron insuficientes para la prueba Χ², sin embargo, los informes de padres y niños/adolescentes fueron consistentes para las categorías de «deteriorado» y «sin cambios» (todos p <0.01). Los resultados completos están disponibles en la Figura 2 a mostrada más abajo. Se informó deterioro de la depresión, ansiedad, irritabilidad, atención e hiperactividad hasta en un 46,5 a 53,6% de los niños y adolescentes, según la edad. Por otro lado, se observaron mejoras en estos dominios hasta en un 11 – 19,6% de los participantes. En el caso de las obsesiones/compulsiones, se observó un deterioro en el 19,7-22,6% de los que respondieron, mientras que se observaron mejoras en el 3-4% de los niños y adolescentes. Cuando se combinaron los datos de todas las edades, el dominio con la tasa más alta de deterioro fue la atención (47,3%), evaluado en niños/adolescentes de 6 a 18 años (consulte la Tabla 2 para ver los resultados completos).
Cambio del estado de salud mental en todos los grupos de diagnóstico de salud mental
Los niños/adolescentes con NO DX experimentaron un deterioro en la depresión, irritabilidad, atención, hiperactividad y obsesión/compulsión de 13,0 a 40,8%, según el dominio. El deterioro de la depresión, irritabilidad, atención e hiperactividad fue mayor en niños y adolescentes con TEA (56,1-66,7%) mientras que el deterioro de la ansiedad y obsesión/compulsión fue mayor entre los niños y adolescentes con diagnósticos de MH + TEA.
Por otro lado, se observaron mejoras en estos dominios hasta en el 0,9–18,8% de los participantes, según la categoría y el dominio de diagnóstico. En la figura 2b mostrada debajo se presenta la prevalencia del cambio en el estado de salud mental, por dominio, en niños con y sin trastornos de salud mental y neurodesarrollo anteriores a COVID.
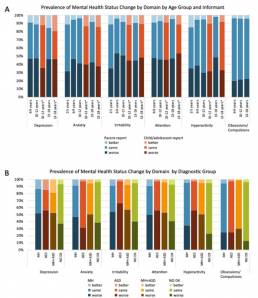
Figura 2. A) Prevalencia del cambio del estado de salud mental in niños/adolescentes entre 2-18 años agrupados por dominio, edad e informante. B) Prevalencia en el cambio de estado de salud mental agrupado por dominio y grupo diagnóstico.
Factores de riesgo y de protección
La consistencia interna de las medidas del informe de los padres de niños de 6 a 18 años sobre el impacto económico de COVID-19 y el estrés del aislamiento social fue aceptable.
Los autores plantearon la hipótesis de que el estrés del aislamiento social y la exposición al COVID-19 tendrían impactos negativos en todos los dominios de la salud mental de niños y adolescentes. El deterioro de la depresión se asoció con la presencia de un diagnóstico psiquiátrico pre-COVID (OR = 2,04, IC 95% 1,45-2,86, p <0,001) y un mayor estrés por aislamiento social (OR = 55,24, IC 95% 24,55-124,30, p < 0,001). El deterioro de la ansiedad se asoció con un mayor estrés por aislamiento social (OR = 54,36, IC del 95%: 25,03-118,03, p <0,001). El deterioro de la irritabilidad se asoció con la etnia/ascendencia europea/norteamericana del niño (OR = 0,58, IC del 95%: 0,42-0,80, p = 0,001), la presencia de un diagnóstico pre-COVID (OR = 2,08, IC del 95%: 1,48-2,92, p <0,001), edad del niño mayor (OR = 0,93, IC del 95% 0,88 a 0,98, p = 0,003) y estrés por aislamiento social (OR = 43,25, IC del 95% 19,59 a 95,46, p <0,001). El deterioro de la atención se asoció con un mayor estrés por aislamiento social (OR = 24,93, IC del 95% 11,76–52,87, p <0,001). El deterioro de la hiperactividad se asoció con ser hombre (OR = 0,72, IC del 95%: 0,52 a 0,99, p = 0,043), tener un diagnóstico psiquiátrico pre-COVID (OR = 2,23, IC del 95%: 1,56 a 3,19, p <0,001), mayor edad del niño (OR = 0,92, IC del 95% 0,88–0,97, p = 0,002) y mayor estrés por aislamiento social (OR = 16,74, IC del 95% 7,94–35,29, p <0,001). El deterioro de la obsesión/compulsión se asoció con tener un diagnóstico psiquiátrico pre-COVID (OR = 1,96, IC 95% 1,30-2,98, p = 0,002), mayores preocupaciones económicas (OR = 2,91, IC 95% 1,45-5,85, p = 0,003) y mayor estrés por aislamiento social (OR = 11,12, IC 95% 5,01-24,70, p <0,001).
La mejoría en la depresión se asoció con tener un diagnóstico psiquiátrico pre-COVID (OR = 3,12, IC del 95%: 1,81 a 5,37, p <0,001) y menos estrés por aislamiento social (OR = 0,16, IC del 95%: 0,04 a 0,56, p = 0,004). La mejora de la ansiedad se asoció con ser mujer (OR = 1,84; IC del 95%: 1,02–3,31, p = 0,041), presencia de un diagnóstico psiquiátrico pre-COVID (OR = 2,42; IC del 95%: 1,17–5,01, p = 0,018), mayores preocupaciones económicas (OR = 5,57, IC 95% 1,64-18,97, p = 0,006) y menor estrés por aislamiento social (OR = 0,12, IC 95% 0,02-0,62, p = 0,011). La mejora en la irritabilidad se asoció con tener un diagnóstico psiquiátrico pre-COVID (OR = 2,13, IC del 95%: 1,10–4,13, p = 0,024). La mejora en la atención se asoció con mayores preocupaciones económicas (OR = 3,97, IC del 95%: 1,29-12,22, p = 0,016). El modelo de mejora de la hiperactividad no fue significativo (p = 0,07), por lo que no se indicaron más predictores. La mejora en la obsesión / compulsión se asoció con la etnia / ascendencia no europea/norteamericana (OR = 2,49, IC 95% 1,18-5,24, p = 0,016) y mayores preocupaciones económicas (OR = 5,20, IC 95% 1,27-21,21, p = 0.022; Tabla 3).
DISCUSIÓN
Este estudio, realizado de dos a cuatro meses después de la implementación de las medidas de emergencia sanitaria por COVID-19, indica que muchos niños y adolescentes en Canadá experimentaron un deterioro en varios dominios de salud mental durante este período de tiempo. Más de dos tercios de los niños y adolescentes experimentaron un deterioro en la salud mental, el cual estuvo fuertemente asociado con el estrés relacionado con el aislamiento social. Estos impactos ocurrieron en ausencia de una exposición significativa a la enfermedad COVID-19 o de preocupaciones económicas entre los participantes del estudio, cuando la prevalencia viral en la comunidad se estimó en 128,6 casos por 100.000, y antes de que se hayan flexibilizado las medidas de confinamiento.
Una proporción significativa de niños y adolescentes experimentó un deterioro de su salud mental, independientemente de la edad, con un 70,2% de los niños de 6 a 18 años y un 66,1% de los niños de 2 a 5 años que experimentaron un deterioro en al menos un dominio. El deterioro de la depresión fue más alto entre los niños de 10 a 12 años, mientras que el deterioro de la ansiedad y la irritabilidad fue más alto entre los niños de 6 a 9 años. El deterioro de la atención, la hiperactividad y la obsesión/compulsión fueron mayores entre los adolescentes de 13 a 18 años. Los niños en edad preescolar tenían menos probabilidades de mostrar un deterioro de la ansiedad, la irritabilidad y la hiperactividad, con las tasas más bajas de deterioro y, a menudo, las tasas más altas de mejora en cada grupo de edad. Esto indica que los impactos de la pandemia en la salud mental son mayores para los niños en edad escolar y potencialmente, de acuerdo con investigaciones anteriores, relacionados con la pérdida de las rutinas diarias de los niños de todas las edades.
En particular, los niños y adolescentes experimentaron impactos en la salud mental relativamente similares a pesar de su historial de salud mental, aunque se observaron algunas variaciones. Se informó que los niños y adolescentes con diagnósticos de TEA tienen el mayor deterioro en depresión, irritabilidad, atención e hiperactividad. Esto puede deberse a varios factores, incluido el cierre de los servicios escolares (incluidos los servicios terapéuticos) para niños con TEA, junto con los desafíos asociados con el aprendizaje en línea, el cierre de los servicios de cuidado de relevo y las interrupciones en las rutinas diarias. Sin embargo, más preocupante, fue la proporción significativa de niños y adolescentes sin problemas conocidos de salud mental o desarrollo neurológico antes de COVID-19 que experimentaron deterioro en varios dominios de salud mental, con un 37-41% experimentando un empeoramiento de la depresión, ansiedad, irritabilidad y atención durante las medidas de emergencia.
Zhou et al. (2020) informaron que el 43,7% y el 37,4% de los niños de 12 a 18 años informaron tasas clínicamente significativas de depresión y ansiedad, respectivamente, en la provincia de Hubei, China, que experimentó una alta carga de COVID-19. En los Estados Unidos, a pesar de los conocidos desafíos con COVID-19, la salud del comportamiento informada por los padres en los niños menores de 18 años solo se informó que fue peor durante la pandemia en el 14,3% de la muestra. Los hallazgos de este estudio concuerdan con un estudio representativo en Alemania que mostró que el cumplimiento de las medidas de emergencia y el aislamiento social son desafíos importantes para los niños y adolescentes. Sin embargo, se evidenció una tasa más alta de deterioro en los dominios de salud mental, con un 17,8% de problemas de salud mental informados por los padres en Alemania en comparación con alrededor del 70% con un deterioro de la salud mental informado por los padres en al menos uno de los seis dominios evaluados en este estudio. La tasa notoriamente más alta de deterioro de la salud mental en los niños y adolescentes canadienses puede deberse a nuestro interés en evaluar múltiples dominios de la salud mental, cualquiera de los cuales puede haber sido afectado en un individuo, como se refleja en las tasas de deterioro relativamente más bajas dentro de cada dominio individual. Estos hallazgos amplían estudios previos en Brasil, China, Alemania, India, Italia, España y los Estados Unidos, al informar sobre el cambio del estado de salud mental en comparación con la salud mental previa al COVID-19 en varios dominios, y sugieren que el impacto generalizado de la pandemia en niños y adolescentes es más probable que sea atribuible a factores distintos a la magnitud de la prevalencia viral y la prevalencia viral de COVID-19 en la comunidad.
En los seis dominios, el mayor estrés del aislamiento social, incluida la cancelación de eventos importantes y la pérdida de interacciones sociales en persona, se asoció fuertemente con el deterioro de la salud mental en todos los dominios. Un menor estrés por aislamiento social se asoció con una mejora en la depresión y ansiedad. Este hallazgo subraya la importancia de la escuela, la recreación, las actividades sociales y los eventos importantes. Una estrategia importante de mitigación para disminuir el deterioro en los seis dominios de la salud mental analizados sería aumentar el acceso a interacciones sociales y recreativas para niños y adolescentes de todas las edades. Al seguir las pautas de salud pública locales, los niños deben recibir apoyo para que asistan a la escuela en persona, participen en deportes y actividades, se reúnan socialmente con sus compañeros, participen en interacciones sociales positivas dentro de la familia y se conecten con familiares y amigos a través de plataformas de video chat.
Los diagnósticos psiquiátricos y del neurodesarrollo previos a COVID-19 se asociaron con mejoras (ansiedad, depresión, irritabilidad) y deterioro (hiperactividad, ansiedad, obsesión/compulsión, depresión, irritabilidad) en la salud mental en todos los dominios, destacando así la heterogeneidad de las respuestas de niños y adolescentes a expectativas sociales y académicas en el hogar y la escuela. Para algunos niños con diagnósticos pre-COVID-19 (p. Ej., Aquellos con ansiedad social o trastornos del aprendizaje), las directivas de quedarse en casa pueden haber proporcionado alivio de las fuentes de estrés o ansiedad, mejorando así sus síntomas de ansiedad o irritabilidad. Para los niños con otros diagnósticos (p. Ej., TEA o TDAH), la pérdida de estructura, consistencia y las interacciones familiares y sociales entre maestro y alumno pueden haber llevado a una mayor irritabilidad y bajo estado de ánimo, potencialmente exacerbado por otros factores como el estrés de los padres secundario a las tareas adicionales de apoyo a los niños con la escolarización.
En conjunto, estos hallazgos sobre las diferencias interindividuales sugieren que los niños con problemas de salud mental preexistentes no son homogéneos con respecto a su respuesta a la pandemia de COVID-19 y pueden experimentar mejoras y deterioros de manera diferente según los perfiles de síntomas específicos dentro de un individuo, en lugar de los diagnósticos. Además, los resultados del estudio hacen énfasis en la necesidad de realizar investigaciones sobre intervenciones efectivas para mitigar el aumento de los síntomas a fin de evitar una mayor angustia durante el regreso a la escuela de algunos niños, así como durante las siguientes oleadas de pandemias. Las estrategias de mitigación para los niños con enfermedades psiquiátricas preexistentes, así como la aparición de nuevos problemas de salud mental, pueden incluir garantizar el acceso continuo a los servicios de salud mental para pacientes ambulatorios, así como el acceso a la atención de salud mental aguda para aquellos que se encuentran en crisis.
Las mayores preocupaciones económicas de COVID se asociaron con mejora en la ansiedad, atención y obsesión/compulsión y también se asociaron con el deterioro de la obsesión/compulsión, lo que indica que puede haber una relación no lineal entre las preocupaciones económicas de COVID y la salud mental infantil. Las preocupaciones económicas de COVID pueden constituir un factor protector en ciertas situaciones, que incluyen: reducción del conflicto entre la vida laboral y personal, la reducción del estrés laboral que implica un alto riesgo de exposición al COVID-19, la recepción de beneficios financieros de emergencia que pueden haber excedido los salarios de empleos mal remunerados, beneficios financieros de emergencia que alivian las limitaciones financieras concomitantes con la pérdida del empleo, o la disponibilidad relativa de padres desempleados versus padres empleados durante las medidas de emergencia. Por ejemplo, los padres que perdieron el empleo pueden haberse sentido menos estresados por el conflicto entre el trabajo y la vida y/o pueden haber tenido más tiempo para pasar con sus hijos, brindándoles apoyo emocional o ayudando con la escuela online, sabiendo que las amenazas financieras inmediatas se estaban mitigando por beneficios económicos de emergencia. En apoyo a esta suposición, una revisión de alcance sistemática reciente demostró que las intervenciones similares a la renta básica universal, al igual que los beneficios financieros de emergencia ofrecidos en Canadá durante la primera ola de la pandemia, tuvieron fuertes efectos positivos en la salud mental de los receptores. Se requieren más análisis, tanto cuantitativos como cualitativos, para determinar las características específicas de la muestra, las condiciones y los posibles mecanismos por los cuales las mayores preocupaciones económicas durante el COVID-19 se asociaron con una mejora en la ansiedad, la atención y la obsesión/compulsión en niños y adolescentes.
CONCLUSIÓN
La mayoría de los niños y adolescentes de este estudio experimentaron un deterioro de su salud mental durante la fase inicial de implementación de las medidas de emergencia. Además, los niños con diagnósticos preexistentes de HM y/o TEA experimentaron cambios en el estado comparables en los dominios en comparación con los niños/adolescentes sin diagnósticos psiquiátricos preexistentes. El predictor más fuerte del deterioro de la salud mental fue el estrés debido al aislamiento social. Se deben alentar los esfuerzos para mantener o adaptar las actividades de los infantiles, en lugar de cancelarlas. Los sectores de las políticas públicas, los sistemas educativos y la salud mental deben equilibrar el riesgo de infección con el deterioro de la salud mental de niños y adolescentes observado en esta primera ola, a medida que se tomen decisiones sobre el reingreso a las escuelas, las actividades recreativas y otras actividades normativas.
Referencia:
Cost, K.T., Crosbie, J., Anagnostou, E. et al. Mostly worse, occasionally better: impact of COVID-19 pandemic on the mental health of Canadian children and adolescents. Eur Child Adolesc Psychiatry (2021).
Ver publicación original aquí:
https://doi.org/10.1007/s00787-021-01744-3
Mayormente peor ocasionalmente mejor rev GS


Evidencia de escape de la variante B.1.351 del SARS-CoV-2 de suero natural e inducido por vacuna
La carrera para producir vacunas contra el SARS-CoV-2 comenzó cuando se publicó la primera secuencia, lo cual constituye la base de las vacunas que se utilizan actualmente en todo el mundo.
Recientemente se han notificado 52 linajes de SARS-CoV-2: B.1.1.7 se identificó por primera vez en el Reino Unido a partir de una muestra obtenida en octubre de 2020; B.1.351 se identificó en octubre de 2020 en Sudáfrica y P.1 en Brasil en diciembre de 2020 (https://www.cogconsortium.uk/wp-content/uploads/2021/01/Report96 2_COG-UK_SARS-CoV-2-Mutations.pdf).
Lo más preocupante son las mutaciones en el dominio de unión al receptor (RBD, de sus siglas en inglés receptor binding domain) de la proteína de pico. El RBD está contenido en la subunidad S1 de spike y es responsable de interactuar con el receptor celular del SARS-CoV-2: la enzima convertidora de angiotensina-2 (ACE2). La superficie de interacción de ACE2 contiene 25 aminoácidos y, debido a su papel crucial en la unión, también es el sitio de unión de muchos anticuerpos neutralizantes. Se cree que el bloqueo de la interacción RBD/ACE2 juega un papel importante en la protección inducida contra la infección por SARS-CoV-2. Las variantes del SARS-CoV-2 antes mencionadas tienen múltiples cambios en la proteína de pico inmunodominante que facilita la entrada de células virales a través del receptor ACE2.
Por tanto, las mutaciones en el sitio de reconocimiento del receptor en la proteína pico son de gran preocupación por su potencial de causar un escape inmune.
La superficie de unión de ACE2 es, hasta cierto punto, el talón de Aquiles del virus, ya que puede ser bloqueado por algunos anticuerpos neutralizantes. Dado que es tan pequeño, amenaza con producir escape inmunológico, ya que pequeños cambios pueden eliminar los anticuerpos neutralizantes, reduciendo la capacidad de inmunidad (natural o adquirida mediante la vacunación) para contener la replicación viral.
Presión selectiva para cambios en la superficie de interacción ACE2 pueden tener dos impulsores completamente separados. En primer lugar, dado que el SARS-CoV-2 ha cruzado recientemente una barrera zoonótica, se puede esperar que la evolución de la superficie de interacción de ACE2 pueda ocurrir para aumentar la afinidad por ACE2 y, por lo tanto, aumentar transmisibilidad viral. Por otro lado, los cambios en la superficie de interacción ACE2 también pueden reducir la protección brindada por una infección o vacunación previa, lo que podría conducir a un escape de la inmunidad preexistente inducida por infecciones naturales o vacunas.
Las tres cepas variantes del SARS-CoV-2 identificadas recientemente han adquirido mutaciones en la superficie interactiva. Todas ellas pueden conducir a un aumento de la transmisibilidad, lo cual fue observado con la variante B.1.1.7 en Reino Unido.
Daming Zhou y colaboradores realizaron un análisis de la estructura y función de la variante B.1.351, utilizando una gran cohorte de muestras de suero de convalecientes y vacunados. Las mutaciones del dominio de unión al receptor proporcionan una unión más estrecha de ACE2 y un escape generalizado del anticuerpo monoclonal, neutralización impulsada en gran medida por E484K. En varios casos, parecería que los pacientes convalecientes y algunos sueros de vacunas ofrecen una protección limitada contra esta variante.
Neutralización de B.1.351 por plasma de convalecencia
Se compraron los títulos de neutralización contra Victoria (SARS-CoV-2 / human / AUS / VIC01 / 2020), una de las primeras cepas relacionadas con SARS-CoV-2, utilizando una prueba de neutralización por reducción de enfoque (FRNT).
Para las primeras 170 muestras convalecientes, los títulos de neutralización contra B.1.351 fueron en promedio 13,3 veces más chicas en comparación con Victoria (p = <0,0001).
Neutralización de B.1.351 por suero vacunado.
Se midió luego la neutralización de Victoria y B.1.351 utilizando suero de vacuna obtenido de
190 personas vacunadas con la vacuna Pfizer-BioNTech BNT162b2 o la vacuna Oxford191 AstraZeneca AZD1222.
Para el suero de la vacuna Pfizer-BioNTech, 196 para B.1.351 fueron 7.6 veces más bajos que los de Victoria (p = <0.0001) y para el suero de la vacuna Oxford-AstraZeneca, los títulos de la media geométrica B.1.351 fueron 9 veces más bajos que para Victoria (p <0,0001). El plasma tomado antes de la primera dosis de la vacuna Oxford-AstraZeneca mostró, como se esperaba, una neutralización mínima o ausente de virus Victoria o B.1.351.
El suero de la vacuna Pfizer-BioNTech indujo títulos de neutralización 3.6 veces más altos contra la cepa Victoria que la vacuna Oxford-AstraZeneca (p = <0,0001).
Mutaciones en el RBD de B.1.351
La triple mutación K417N, E484R y N501Y es característica del RBD de B.1.351. Estos residuos están situados dentro de la huella de ACE2 (ver Figura 1) y optimizan la afinidad por ACE2, por lo cual confieren una mayor afinidad por el receptor. Para investigar este efecto, se midió la cinética
de unión de ACE2 soluble a RBD recombinante mediante interferometría de biocapa (BLI). Como era de esperar, la afinidad por B.1.351 RBD fue 19 veces mayor que para el RBD de Victoria y 2,7 veces más alto que para B.1.1.7.

Figura 1
Neutralización de B.1.351 por un gran panel de anticuerpos monoclonales
Se ha producido y caracterizado un conjunto de 377 anticuerpos monoclonales humanos dirigidos a la proteína de pico, obtenida de muestras de convalecientes de pacientes infectados durante la primera ola de SARS-CoV-2 en el Reino Unido antes de junio de 2020, por lo tanto, no inducida en la respuesta a la infección con cepas recientes de SARS-CoV-2.
Los efectos sobre la neutralización de mAb fueron severos, 14/20 anticuerpos tuvieron una caída de más de 10 veces en los títulos de neutralización, y la mayoría de estos muestran una inactivación completa de la actividad. Esto se debe a los roles clave de K417, E484 y N501, en particular E484 en el reconocimiento de anticuerpos de la superficie de interacción ACE2 del RBD.
Curiosamente, el único y potente anticuerpo de unión a dominio N- terminal (NTD) incluido en estos análisis, mAb 159, también mostró una eliminación completa de la actividad contra B1.1351. Como puede verse en la Figura 2, el bucle 246-253 del RBD interactúa con la cadena pesada del mAb 159. La supresión 242-244 indudablemente alterará la presentación de este bucle, comprometiendo la unión a estos mAb. Se ha informado de la vinculación en este llamado «supersitio» como de posible relevancia terapéutica.
Tanto B.1.1.7 como B.1.351 y P.1 poseen deleciones o cambios sistemáticos en el NTD.
Dado que estas características convergentes pueden haber surgido antes de una fuerte presión de la respuesta selectiva del anticuerpo, parece probable que todavía haya un impulsor biológico subyacente por descubrir, como el aumento de la unión al receptor y el potencial aumento de la transmisibilidad impartida por mutaciones en el RBD.

Figura 2
Discusión
Los coronavirus son virus de ARN de cadena positiva y, aunque la ARN polimerasa posee una capacidad limitada de corrección de pruebas, son intrínsecamente propensos a cambios mutacionales.
La evolución del SARS-CoV-2 a partir de una probable introducción zoonótica de un solo punto en Wuhan en noviembre/diciembre 2019, ha sido ampliamente anticipado y condujo al establecimiento de vigilancia de secuencias virales como COG-UK en el Reino Unido.
La reciente aparición de tres cepas de SARS-CoV-2 (B.1.1.7, B1.351 y P.1), que causar una mayor transmisibilidad, se ha producido de forma independiente en el Reino Unido, Sudáfrica y Brasil, donde se han convertido rápidamente en cepas dominantes y ahora se están extendiendo a nivel mundial.
Si bien las secuencias son marcadamente diferentes, cada una con 9-12 cambios, todas ellas involucran la superficie de interacción ACE2 del RBD. A su vez, comparten la eliminación en el NTD, los cuales interrumpirán el sitio de unión de anticuerpos anti-NTD neutralizantes, causando el fracaso en la neutralización de mAb anti-NTD.
No se conoce cuánta mutación adicional en el RBD podrán soportar estos anticuerpos conocidos, pero el uso de combinaciones de anticuerpos que protejen contra variantes virales
parece ser una estrategia acertada. De todos modos, debe reconocerse que el uso de terapia o profilaxis con anticuerpos monoclonales, particularmente durante períodos prolongados en individuos inmunodeprimidos con infección crónica, es probable que impulsen la aparición de mutaciones, con la consecuente generación de resistencia.
En este estudio, se demostró que la cepa B.1.351 CoV-2 es mucho más difícil de neutralizar que las cepas parentales. En suero de convalecencia los títulos de neutralización se reducen 13,3 veces para B.1.351 en comparación con la cepa Victoria. Títulos de neutralización para las vacunas Oxford-AstraZeneca y Pfizer se redujeron de manera similar con B.1.351 en 9 y 7,6 veces, respectivamente. Tanto las vacunas de Oxford-AstraZeneca como la de Pfizer muestran una eficacia inicial sustancial después de una sola dosis de vacuna.
Lo que está impulsando la evolución de B.1.351 es difícil de desentrañar: por un lado, se ve un aumento de 20 veces en la afinidad por ACE2 en comparación con el RBD de Wuhan, lo cual puede influir en la transmisibilidad. Por otro lado, el escape inmune sustancial de anticuerpos por B.1.351
probablemente desempeña un papel en países como Sudáfrica, donde las tasas de infección previa son relativamente altas (mayores a 30%). La compensación de una mayor afinidad ACE2 y
la transmisibilidad contra el escape inmune es probablemente compleja, a medida que aumenta la inmunidad de la población debido a la vacunación y la infección natural. La capacidad de generar variantes de RBD de afinidad alta para ACE2 y una mayor afinidad que casi todos los anticuerpos monoclonales descritos hasta la fecha, es motivo de preocupación.
En resumen, la reciente aparición de múltiples cepas variantes de SARS-CoV-2 ha interrumpido
la confianza en torno a si la generación actual de vacunas proporcionará protección contra las infecciones. Esto a provocado prisa por comprender las consecuencias de estos cambios y ha estimulado un impulso para desarrollar nuevas vacunas adaptadas a las variantes, particularmente incorporando la mutación E484K.
Cómo responden las personas previamente infectadas o vacunadas a estas nuevas variantes será objeto de un intenso estudio durante los próximos meses, ya que existe un reconocimiento general de que el problema actual no ha terminado. Sin embargo, incluso si las respuestas de anticuerpos
a las nuevas variantes no pueden prevenir la infección, pueden moderar la gravedad. Además,
las respuestas de las células T 470 al pico pueden no verse interrumpidas por los cambios mutacionales y ser capaces de limitar la diseminación de la enfermedad al tracto respiratorio inferior, previniendo enfermedades graves. Es necesario implementar sistemas de vigilancia intensiva para monitorear la aparición de nuevas variantes, así como trabajar en vacunas de segunda e incluso de tercera generación dirigidas a las nuevas variantes del virus.
Referencias:
1) Zhou, D., Dejnirattisai, W., Supasa, P., Liu, C., Mentzer, A.J., Ginn, H.M., Zhao, Y., Duyvesteyn, H.M.E., Tuekprakhon, A., Nutalai, R., Wang, B., Paesen, G.C., Lopez-Camacho, C., Slon-Campos, J., Hallis, B., Coombes, N., Bewley, K., Charlton, S., Walter, T.S., Skelly, D., Lumley, S.F., Dold, C., Levin, R., Dong, T., Pollard, A.J., Knight, J.C., Crook, D., Lambe, T., Clutterbuck, E., Bibi, S., Flaxman, A., Bittaye, M., Belij-Rammerstorfer, S., Gilbert, S., James, W., Carroll, M.W., Klenerman, P., Barnes, E., Dunachie, S.J., Fry, E.E., Mongkolspaya, J., Ren, J., Stuart, D.I., Screaton, G.R., Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine induced sera, Cell (2021), doi:
https://doi.org/10.1016/j.cell.2021.02.037
Nota_Evidencia variante B.1.351


Estrategias de vacunas contra el SARS-CoV-2: Candidatos de fase 3
NPJ Vaccines. 2021 Feb 22;6(1):28.
doi: 10.1038/s41541-021-00292-w. PMID: 33619260.
https://www.nature.com/articles/s41541-021-00292-w
SARS-CoV-2 es un virus de ARN que pertenece a la familia Coronaviridae y causa la enfermedad COVID-19. El virus se habría originado en China, donde fue secuenciado inicialmente y se extendió rápidamente por todo el mundo mediante viajeros y turistas, convirtiéndose en una pandemia que, hasta el 5 de enero de 2021, causó más de 1.866.000 muertes. Una vacuna eficaz contra esta enfermedad será fundamental para reducir la morbilidad y la mortalidad.
El genoma del virus codifica varias proteínas estructurales y no estructurales, incluidas las proteínas Spike (S), de envoltura (E), de membrana (M) y nucleocápside (N). La mayoría de las vacunas candidatas para COVID-19 que emplean la administración de antígenos virales o secuencias de genes virales tienen como objetivo inducir anticuerpos neutralizantes contra la proteína Spike (S), previniendo la captación a través del receptor ACE2 humano y, por lo tanto, bloqueando la infección. Sin embargo, un número creciente de bibliografía ha destacado la importancia de las respuestas celulares en la recuperación de los pacientes con COVID-19, promoviendo así, no solo el uso de estrategias de vacuna que favorecen la inducción de respuestas mediadas por células T, sino también el screening de su producción en participantes de ensayos clínicos. Por otro lado, las estrategias que utilizan virus completo, ya sea atenuado o inactivado, aspiran a inducir una respuesta policlonal más amplia y heteróloga contra varios antígenos virales.
Desde la publicación de la secuencia del genoma del SARS-CoV-2, el 11 de enero de 2020, un esfuerzo de una velocidad y magnitud sin precedentes se propuso desarrollar una vacuna contra la enfermedad. Los avances recientes en el campo han hecho posible la emisión de autorizaciones de uso de emergencia (EUA) por parte de varias agencias reguladoras de medicamentos nacionales e internacionales para diferentes candidatos a vacunas contra el SARS-CoV-2 en menos de un año desde que se lanzó la secuencia del genoma del virus.
Una vacuna ideal contra el SARS-CoV-2 debe cumplir los siguientes requisitos: proteger no solo de enfermedades graves sino también contrarrestar la infección en todas las poblaciones vacunadas, incluidas las personas menos inmunodeprimidas; provocar respuestas inmunitarias de memoria a largo plazo después de un número mínimo de inmunizaciones o dosis de refuerzo; la empresa de fabricación debería poder aumentar la producción para producir miles de millones de dosis al año y tener el potencial de hacerla fácilmente accesible para las campañas de vacunación en todo el mundo a un costo asequible y en un tiempo limitado.
Cuatro iniciativas diferentes se encuentran entre las fuentes esenciales de financiación que permitieron el desarrollo de varias vacunas candidatas al SARS-CoV-2. Una de las primeras fuentes de financiación fue la Coalition for Epidemic Preparedness Innovations (CEPI), una asociación mundial sin fines de lucro que se dedica a proporcionar fondos para vacunas para detener las epidemias emergentes. Otra fuente vital de fondos provino de la Autoridad de Investigación y Desarrollo Biomédico Avanzado (BARDA), que ha asignado varios millones de dólares a los principales candidatos a vacunas y otros tratamientos prometedores de COVID-19. El programa de vacunas de la Unión Europea tiene en marcha un esfuerzo conjunto para comprar vacunas para los países de la UE. Esta entidad ya ha firmado contratos con seis desarrolladores de vacunas, incluidos Pfizer y BioNTech, Sanofi-GSK, Curevac, AstraZeneca y la Universidad de Oxford, Johnson & Johnson y Moderna. Más recientemente, la Operación Warp Speed del gobierno de EE. UU. invirtió más de mil millones de dólares para financiar el desarrollo de 8 candidatos de vacuna líderes para acelerar su evaluación, aprobación y fabricación para EE. UU. Por último, Gavi, una alianza de vacunas de acceso global, CEPI y la Organización Mundial de la Salud (OMS) han lanzado la iniciativa COVAX (Acceso a la vacuna contra el coronavirus) para garantizar el acceso equitativo de las vacunas del SARSCoV-2 a países no autofinanciados que carecen de recursos.
Según la OMS, al 5 de enero de 2021, hay 63 vacunas candidatas en ensayos clínicos en humanos y más de 172 candidatas en desarrollo preclínico en todo el mundo. Entre las 60 vacunas evaluadas clínicamente, se encontraron 13 candidatos líderes que ya están realizando o ingresando a ensayos clínicos de Fase 3. Las tecnologías de plataforma han sido empleadas por diferentes grupos de investigación para desarrollar sus candidatos a vacunas, en donde es de esperarse que los primeros candidatos a entrar en los ensayos clínicos de Fase 3 estén utilizando estrategias de despliegue rápido, a saber, plataformas de ácido nucleico, plataformas de vectores virales no replicantes, virus inactivados o vacunas de subunidades recombinantes. Otras estrategias tradicionales de desarrollo de vacunas, como las vacunas con virus atenuados, requieren largos procesos de cultivo celular para lograr cepas atenuadas. Es muy posible que las vacunas contra el SARS-CoV-2 de segunda generación demuestren la capacidad de provocar respuestas de memoria más sólidas y más largas con una sola inmunización.
Resumen de las estrategias utilizadas para el desarrollo y la administración de vacunas
Hay dos objetivos principales que cualquier estrategia de vacuna debe lograr: la seguridad de la vacuna y la producción de respuestas inmunitarias adaptativas sólidas que conducen a protección a largo plazo contra varias cepas del patógeno con -idealmente- una dosis de la vacuna.
Un creciente cuerpo de evidencia sugiere que las respuestas mediadas por células T contra el SARS-CoV-2 son extremadamente importantes y más duradera que la inmunidad de células B. Por lo tanto, las estrategias de vacuna que inducen fuertes respuestas celulares además de la inmunidad humoral presentan una ventaja significativa en la presente pandemia.
Es así, que existen varias estrategias (ver Figura 1):
A) Vacunas de patógenos atenuados: Las estrategias de vacuna de patógenos vivos atenuados consisten en administrar una forma debilitada de patógenos vivos. Los pases prolongados de cultivos celulares en líneas celulares no humanas o animales disminuyen la virulencia del patógeno. Este tipo de vacunas generalmente provoca respuestas inmunes de memoria robustas y de largo plazo después de una sola dosis.
B) Vacunas de patógenos inactivados: Las vacunas de patógenos inactivados contienen patógenos completos que han sido sometidos a inactivación por tratamiento térmico o químico.
C) Vacunas de subunidad: Las vacunas de la subunidad se preparan a partir de la purificación de antígenos de patógenos replicados en cultivos celulares o de antígenos expresados de forma recombinante. Estas vacunas comúnmente requieren la adición de adyuvantes para enviar señales de peligro a las células presentadoras de antígenos y provocar respuestas inmunes robustas.
D) Vacunas de partículas similares a virus: Las partículas similares a virus se pueden autoensamblar y liberar a partir de células de levadura recombinantes u otros sistemas de expresión como el sistema de expresión del virus vaccinia o incluso plantas de tabaco transfectadas con el virus del mosaico del tabaco.
E) Vacunas con vectores virales: Las vacunas de vectores virales utilizan una plataforma adenoviral o contra el sarampión manipulada genéticamente para expresar un antígeno extraño que comúnmente da como resultado una respuesta celular y humoral sólida.
F) y G) Vacunas de ácido nucleico: Las vacunas de ácido nucleico (ADN y ARNm) son muy rápidas de producir, pero no se probaron como estrategias exitosas de vacunas humanas. El ácido nucleico que codifica una proteína inmunogénica del patógeno, una vez administrado, es capturado por las células presentadoras de antígeno que lo utilizan para expresar y presentar el antígeno. Se prevé que estas vacunas tengan problemas menores de seguridad, ya que el ácido nucleico se degrada rápidamente dentro del cuerpo humano.

Fig 1: Resumen de las estrategias utilizadas para el desarrollo y la administración de vacunas
Candidatos actuales para la vacuna contra el SARS-CoV-2 en Fase 3 de la evaluación clínica
Vacunas de ácido nucleico
Vacunas de ARNm
ARNm-1273 (Moderna / US NIAID): Moderna Therapeutics, con sede en Boston, se asoció con el Instituto Nacional de Alergias y Enfermedades Infecciosas (NIAID) para producir la primera vacuna candidata que entró en ensayos clínicos en 63 días después de la secuenciación del genoma del SARS- CoV-2. La vacuna se basa en una molécula de ARNm que contiene la información para la síntesis de la forma de prefusión estabilizada de la proteína SARS-CoV-2 Spike (S) encapsulada en un vector de nanopartículas lipídicas (LNP) que mejora la captación por las células inmunes del huésped. El ARNm administrado utiliza la maquinaria de transcripción y traducción de la célula huésped para producir el antígeno viral que luego se presenta en los linfocitos T y también es reconocido directamente por los linfocitos B del huésped, iniciando así una respuesta inmune adaptativa dirigida contra la proteína del virus.
La dosificación de los primeros voluntarios con ARNm-1273 comenzó el 16 de marzo en 45 voluntarios sanos de entre 18 y 55 años de edad que recibieron tres dosis diferentes, a saber, 25 μg, 100 μg y 250 μg de ARN en forma de estimulación inicial. La segunda vacuna se administró 28 días después de la primera. El informe del ensayo de fase 1 describió una respuesta humoral dependiente de la dosis y la producción de anticuerpos neutralizantes en concentraciones similares a los detectados en sueros convalecientes de COVID-19. Además, las dos dosis más bajas provocaron una fuerte respuesta de los linfocitos T CD4 + con una expresión mínima de citocinas T helper 2 (Th2) que resultaron perjudiciales durante los esfuerzos de desarrollo de vacunas contra el SARS y el MERS (Síndrome respiratorio de Oriente Medio). Por otro lado, las respuestas de los linfocitos T CD8 + solo fueron provocadas por la dosis de 100 μg. El ARNm-1273 fue generalmente bien tolerado y no se informaron efectos adversos de la etapa 4 (incapacitante o potencialmente letal). Los eventos adversos, que comprendieron principalmente mialgia, fatiga, dolor de cabeza, escalofríos y dolor alrededor del lugar de la inyección, fueron más frecuentes después de la segunda inmunización y fueron más prominentes en el grupo de dosis alta (250 μg).
Recientemente se publicaron resultados adicionales de un pequeño estudio de fase 1 en 40 adultos mayores, que se dividieron en dos grupos de edad (56-70 años o ≥71 años). A los participantes se les administraron dos dosis de 25 μg o 100 μg de ARNm-1273 que previamente habían demostrado exhibir un perfil de tolerancia más alto. El perfil inmunogénico de ambos grupos de edad fue bastante similar al informado en el grupo de 18 a 55, lo que sugiere que esta estrategia de vacunación podría ser igualmente inmunogénica en los grupos de edad más vulnerables y generalmente menos inmunocompetentes. Se descubrió que la dosis de 100 μg era más inmunogénica, lo que respalda su uso en un ensayo de vacuna de Fase 3.
Después de completar un ensayo de vacunación de Fase 2 en 300 adultos jóvenes y 300 adultos mayores a los que se les administraron las dosis de 25 o 100 μg, el ARNm-1273 entró en un ensayo de eficacia de Fase 3 el 27 de julio (Identificador de ensayo clínico: NCT04470427). Este ensayo implica la inscripción de 30.000 participantes en los EE. UU., la mitad de los cuales recibirá 2 dosis de 100 μg de ARNm-1273 y la otra mitad un placebo en un estudio aleatorizado doble ciego controlado con placebo. El criterio de valoración principal del estudio es la prevención del COVID-19 sintomático con criterios de valoración secundarios que incluyen la prevención de la infección por SARS-CoV-2 y la prevención de la hospitalización por COVID-19.
El 18 de noviembre, Moderna anunció que su candidata a vacuna alcanzó su criterio de valoración principal de eficacia después de revisar el primer análisis intermedio de su estudio clínico de Fase 3. Este análisis inicial, establecido 14 días después de la segunda vacunación, incluye 95 casos confirmados de COVID-19 entre los participantes, 90 de los cuales pertenecían al grupo que recibió el placebo y 5 al grupo que recibió ARNm-1273. Los cálculos de división de casos revelan una eficacia inicial del 94,5% de la vacuna candidata. Otra observación importante fue que, de los 11 casos graves analizados, ninguno había ocurrido en el grupo vacunado con ARNm-1273, lo que sugiere que posiblemente el ARNm-1273 protege del COVID-19 grave, aunque se necesitan más datos para establecer la certeza. En cuanto al perfil de seguridad del ARNm-1273, se informaron principalmente eventos leves a moderados. Los efectos adversos graves más frecuentes fueron dolor en el lugar de la inyección después de la primera dosis (2,7%) y fatiga (9,7%), mialgia (8,9%), artralgia (5,2%), dolor de cabeza (4,5%), dolor (4,1%). y enrojecimiento en el lugar de la inyección (2,0%) después de la segunda dosis. En general, estos efectos se describieron como de corta duración.
En un segundo comunicado de prensa nuevamente el 16 de noviembre, Moderna reveló que después de pruebas adicionales, se encontró que su vacuna candidata se mantuvo estable durante 30 días entre 2 ° y 8 ° C, hasta por 6 meses a -20 ° C y por hasta 12 h a temperatura ambiente, abordando así uno de los mayores desafíos que presentan las vacunas de ARNm que es la logística de su distribución en áreas rurales y la necesidad de refrigeradores especializados.
El 30 de noviembre, Moderna anunció los resultados del análisis de eficacia principal de su estudio de Fase 3 que involucra 196 casos de COVID-19 confirmado. Entre los 30 individuos que desarrollaron una enfermedad grave, ninguno había sido inmunizado con la vacuna candidata, lo que sugiere que el ARNm-1273 protege fuertemente contra la enfermedad grave. Además, de los 196 casos confirmados de COVID-19, solo 11 pertenecían al brazo de ARNm-1273 del estudio, lo que arroja una estimación puntual de la eficacia de la vacuna del 94,1% 2 semanas después de la segunda dosis. Se informó que la eficacia era constante en todas las edades, razas y etnias, mientras que no se informaron diferencias en el perfil de seguridad del candidato publicado anteriormente. Por lo tanto, la compañía alcanzó los dos puntos finales de 151 casos confirmados y un seguimiento promedio de dos meses de los participantes establecidos en el diseño del estudio y el 18 de diciembre, la Administración de Alimentos y Medicamentos (FDA) de EE. UU. otorgó la EUA de la vacuna en individuos mayores de 18 años de edad, seguido Autoridad Sanitaria de Canadá el 23 de diciembre. El 30 de diciembre, los resultados de seguridad y eficacia del ensayo de Fase 3 del ARNm-1273 se publicaron en el New England Journal of Medicine, confirmando el perfil de seguridad y eficacia del 94,1% de la vacuna candidata. El 4 de enero de 2021, Israel también aprobó la vacuna candidata de Moderna y, posteriormente, la Agencia Europea de Medicamentos (EMA) recomendó la autorización de la vacuna.
ARNm-BNT162b2 / Comirnaty (Pfizer / BioNTech / Fosun Pharma): La segunda plataforma de ARNm candidata es BNT162b2 desarrollada por Pfizer en colaboración con BioNTech (abreviatura de Nuevas Tecnologías Biofarmacéuticas) con sede en Alemania y Fosun Pharma, con sede en Shangai. BioNTech desarrolló y probó inicialmente cuatro candidatos a vacunas modificadas basadas en ARNm (modRNA) diseñadas para administrarse en dos vacunaciones con 3 semanas de diferencia y, tras la inserción en el citoplasma de la célula huésped, instruye a las células inmunitarias para que realicen varias copias de la proteína Spike de SARS-CoV-2 de longitud completa.
Los resultados de los ensayos clínicos aleatorizados controlados con placebo de Fase 1 mostraron que BNT162b2 genera efectos secundarios mínimos tanto en participantes más jóvenes (18-55 años) como mayores (65-85 años). Además, se evaluaron dos candidatos diferentes en estos ensayos, a saber, BNT162b1 y BNT162b2. Ambos candidatos indujeron la producción de concentraciones de anticuerpos neutralizantes dependientes de las dosis igualmente altas contra el SARS-CoV-2 en los participantes inoculados. De hecho, las concentraciones de anticuerpos neutralizantes fueron superiores o iguales a los de los sueros convalecientes del SARS-CoV-2. Sin embargo, BNT162b2 demostró menos reactogenicidad sistémica en adultos mayores y, por lo tanto, la dosis de 30 μg de este candidato fue seleccionada para estudios de eficacia de Fase 2/3 a gran escala. Las respuestas de linfocitos T no se informaron inicialmente para este candidato específico, pero en base a un estudio previo sobre la inmunogenicidad de BNT162b1 se espera una activación significativa de células T CD8 + específicas y poblaciones sesgadas de Th1 CD4 +. De hecho, se descubrió que una inmunización de dos dosis con 30 µg / dosis de BNT162b1 inducía concentraciones elevadas de anticuerpos neutralizantes anti-SARS-CoV-2 y respuestas de linfocitos T Th1 y CD8 + específicas para el paciente. Se realizarán ensayos controlados con placebo aleatorizados de seguridad y eficacia de Fase 2/3 en 43488 voluntarios, incluidos individuos con afecciones crónicas subyacentes y diferentes antecedentes genéticos (Identificador de ensayo clínico: NCT04368728).
El criterio de valoración principal del ensayo es la prevención de COVID-19, y los criterios de valoración secundarios incluyen la prevención de COVID-19 grave y la prevención de la infección por SARS-CoV-2. El ensayo está diseñado para llevarse a cabo en 166 sitios de investigación clínica en todo el mundo en al menos 3 continentes diferentes. La capacidad de fabricación de Pfizer permitirá un suministro global de hasta 50 millones de dosis para fines de 2020 y 1.300 millones de dosis para fines de 2021 si su candidata a vacuna logra la autorización. En un comunicado de prensa emitido el 18 de noviembre, Pfizer y BioNTech anunciaron que los datos del análisis intermedio final sugieren que su candidato demostró una eficacia del 95% contra COVID-19 una semana después de administrar ambas inmunizaciones. Estos datos se basan en la evaluación de 43.448 participantes, 170 de los cuales desarrollaron COVID-19 en la ventana de evaluación. Entre ellos, 162 pertenecen al grupo placebo y 8 al grupo que fue inmunizado con la vacuna candidata. Según el comunicado de prensa, la eficacia de la vacuna fue constante según la edad, el sexo, la raza y la etnia. Además, la eficacia observada en participantes vacunados mayores de 65 años fue superior al 94%. Con respecto al perfil de seguridad de BNT162b2, la vacuna candidata fue bien tolerada en todas las poblaciones ya que no se informaron problemas de seguridad graves. Los eventos de Grado 3 más frecuentes fueron fatiga presentada en 3.8% y dolor de cabeza en 2.0% de los participantes vacunados. Según estos resultados, la empresa considera que se ha cumplido el hito de datos de seguridad exigido por la FDA de EE. UU. para la EUA y el 20 de noviembre, Pfizer y BioNtech se convirtieron en las primeras empresas que presentaron una solicitud de EUA de una vacuna contra el SARS-CoV-2 a la FDA. El 2 de diciembre, BNT162b2 recibió la EUA de la Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA), el regulador de medicamentos del Reino Unido y la administración de vacunas comenzó el 8 de diciembre en el Reino Unido, seguido de una autorización otorgada por la agencia reguladora de medicamentos canadiense el 9 de diciembre. El 11 de diciembre, la FDA otorgó la EUA para BNT162b2 en individuos mayores de 16 años, mientras que la EMA y la Comisión Europea aprobaron la vacuna para individuos mayores de 16 años que residan en cualquiera de los 27 estados miembros de la UE el 21 de diciembre.
Desde entonces, varios otros países han otorgado la EUA para BNT162b2, entre ellos Argentina, Chile, Ecuador, Costa Rica, México, Panamá, Kuwait y Singapur. Suiza, Bahréin y Arabia Saudita también han autorizado la vacuna, y el 31 de diciembre la OMS otorgó la validación de emergencia a BNT162b2, permitiendo así a varios países acelerar los procesos de autorización, importación y distribución de este candidato.
El mismo día se publicaron los resultados de seguridad y eficacia del ensayo clínico BNT162b2 Fase 3 en el New England Journal of Medicine. Además de la confirmación de los 8 casos de COVID-19 entre los participantes asignados a recibir BNT162b2 y 162 casos entre los asignados al placebo se reveló un hallazgo adicional; en 10 casos notificados de COVID-19 grave que se manifestaron después de que los participantes habían recibido la primera dosis, 9 ocurrieron en el grupo de placebo y solo 1 en un receptor de BNT162b2, lo que sugiere que BNT162b2 protege adicionalmente del COVID-19 grave.
Vacunas de ADN
INO-4800 (Inovio / International Vaccine Institute): Aunque la empresa Inovio, con sede en Pensilvania, aún no ha entrado oficialmente en los ensayos de Fase 3, su candidata es la vacuna de ADN del SARS-CoV-2 más avanzada hasta el momento. Inovio Pharmaceuticals ha desarrollado varias vacunas experimentales basadas en ADN que se administran por vía intradérmica con la ayuda de un dispositivo portátil llamado «Cellectra 2000» que emite un pequeño pulso eléctrico que permite una absorción celular y nuclear eficiente de las moléculas de ADN a través de un mecanismo de electroporación. Su candidato es una vacuna de dos dosis.
El 30 de junio, un anuncio de la compañía reveló datos provisionales de un ensayo de Fase 1 en 36 voluntarios de 18 a 50 años de edad que recibieron dos dosis de 1.0 mg o 2.0 mg de INO-4800 con cuatro semanas de diferencia. Estos resultados se publicaron el 23 de diciembre y, según el artículo, no se informaron efectos adversos graves y 34 de los 36 participantes presentaron fuertes respuestas humorales en los grupos de 1,0 mg y 2,0 mg. Además, el 78% de los participantes en el brazo de 1,0 mg y el 84% de los participantes en el brazo de 2,0 mg generaron anticuerpos neutralizantes según lo evaluado por un ensayo de neutralización de cepas SARS-CoV-2 / Australia / VIC01 / 2020. Finalmente, el 74% y el 100% de los participantes inmunizados con 1,0 mg y 2,0 mg, respectivamente, desarrollaron fuertes respuestas de células T Th1 y CD8 +.
El 28 de septiembre, Inovio anunció que la FDA había suspendido parcialmente los ensayos clínicos en Fase 2/3 planificados de la vacuna candidata debido a preguntas sobre el diseño y uso de ‘Cellectra 2000’. El 16 de noviembre, Inovio dijo que el FDA les había dado permiso para seguir adelante con su ensayo de Fase 2/3 llamado INNOVATE65 (Identificador de ensayo clínico: NCT04642638). El brazo de Fase 2 de este estudio se llevará a cabo con 400 voluntarios que recibirán dos dosis por vía intradérmica de 1.0 o 2.0 mg de INO-4800 o un placebo con un intervalo de 4 semanas. El segmento de Fase 3 del estudio involucrará a 6.178 voluntarios que recibirán dosis determinadas por los resultados de seguridad e inmunogenicidad obtenidos del segmento de Fase 2.
Vacunas de vectores virales con replicación defectuosa
Ad5-nCoV (CanSino Biological / Instituto de Biotecnología de Beijing / Academia de Ciencias Médicas Militares): La empresa china CanSino Biologics, en colaboración con el Instituto de Biología de la Academia de Ciencias Médicas Militares de China, desarrolló un candidato que utiliza el vector de adenovirus humano serotipo 5 (Ad5) para entregar la información que codifica la proteína S de longitud completa del SARS-CoV-2 en las células huésped. Ad5 es el principal serotipo adenoviral en humanos, lo que significa que un porcentaje significativo de individuos puede tener contacto reciente y, por lo tanto, inmunidad preexistente contra el vector viral que también podría obstaculizar respuestas inmunes robustas contra el antígeno presentado.
Los datos preliminares de seguridad e inmunogenicidad de Fase 1 obtenidos de 108 participantes entre 18 y 60 años que recibieron dosis bajas, medias y altas de Ad5-nCoV se publicaron el 22 de mayo. Se encontró que las dos dosis más bajas de 5 × 1010 y 1 × 1011 partículas virales tenían un perfil de inmunogenicidad y seguridad aceptable y se seleccionaron para un ensayo de Fase 2.
Los resultados del ensayo de Fase 2, doble ciego, aleatorizado y controlado con placebo también se publicaron el 20 de julio. Se aplicó cualquiera de las dos dosis seleccionadas de Ad5-nCoV o el placebo a un total de 508 voluntarios elegibles de 18 a 83 años de edad. Ambos grupos de dosis provocaron anticuerpos anti-RBD en más del 95% de los participantes el día 28 después de la inmunización y concentraciones de anticuerpos neutralizantes contra el SARS-CoV-2 vivo. Además, alrededor del 90% de los participantes vacunados de ambos grupos demostraron activación de respuestas específicas de células T como lo demuestra el ensayo ELISpot de interferón-γ. El 72% y el 74% de los participantes en los grupos de dosis más alta y más baja informaron reacciones adversas leves, respectivamente. Se documentaron reacciones adversas graves en menos del 10% de los participantes de cada grupo y no se informaron reacciones adversas serias. A raíz de estos resultados, se seleccionó el régimen de una única inmunización de 5 × 1010 partículas virales para proceder en los ensayos de eficacia de Fase 3 que evalúan la protección contra la incidencia de COVID-19 grave con la inscripción de 40.000 voluntarios en Arabia Saudita, Rusia y Pakistán. (Identificador de ensayo clínico: NCT04526990). Mientras tanto, el 25 de junio, la Comisión Militar Central de China anunció que Ad5-nCoV recibió la aprobación para su uso en la adquisición y análisis militar en ausencia de los resultados del ensayo de Fase 3.
AZD1222 (AstraZeneca / Universidad de Oxford): La vacuna vectorizada viral de la Universidad de Oxford y AstraZeneca representa otro candidato. Fue uno de los primeros en comenzar los ensayos clínicos y el único que utilizó una plataforma debilitada de adenovirus de chimpancé (ChAdOx1) para eludir el problema de la inmunidad preexistente contra el vector, ya que muy pocos humanos, si es que hay alguno, tendrían un contacto previo con un virus simio. El vector ChAdOx1 se ha diseñado para incluir la información que codifica la proteína Spike del SARS-CoV-2 de tipo salvaje.
Los primeros resultados de un estudio de control multicéntrico, aleatorizado, simple ciego de fase 1/2 de 1090 voluntarios adultos sanos de entre 18 y 55 años se informaron el 20 de julio. Una proteína meningocócica comparada con una vacuna conjugada meningocócica (MenACWY) sirvió como control en este estudio. Los participantes recibieron una o dos dosis que contenían 5 × 10(10) partículas virales con 4 semanas de diferencia o la vacuna meningocócica de control. El perfil de seguridad de la vacuna se caracterizó como aceptable, mientras que la vacunación homóloga provocó respuestas neutralizantes contra el SARS-CoV-2 en todos los participantes, pero no tuvo efectos adicionales sobre las respuestas adaptativas celulares.
Se están llevando a cabo ensayos de Fase 3 de eficacia y seguridad con el régimen de dos dosis (Identificador de ensayo clínico: NCT04516746) en más de 30.000 personas en los EE. UU., India (Identificador de ensayo clínico: CTRI / 2020/08/027170), Brasil (Identificador del ensayo: ISRCTN89951424), Rusia (Identificador del ensayo clínico: NCT04540393) y Sudáfrica. El 6 de septiembre, un evento adverso grave (AAG) presentado en un paciente inscrito en estudios de Fase 3 detuvo temporalmente los ensayos que se reanudaron en la mayoría de los países, incluido el Reino Unido, pero no en los EE. UU. Esa fue la segunda pausa temporal para un estudio AZD1222, ya que en julio el ensayo se detuvo durante varios días después de que un participante desarrollara síntomas neurológicos graves. Posteriormente se concluyó que estos síntomas se debían a un caso de esclerosis múltiple no diagnosticado previamente que no estaba relacionado con la vacuna. Los criterios de valoración primarios del estudio de Fase 3 se centran en la prevención de COVID-19 y el perfil de reactogenicidad y tolerancia del candidato.
El 23 de noviembre, la Universidad de Oxford y AstraZeneca emitieron comunicados de prensa separados presentando los resultados provisionales de la Fase 3 de su vacuna candidata. Estos resultados se publicaron el 8 de diciembre en Lancet en un análisis de seguridad y eficacia de cuatro ensayos llevados a cabo en Brasil, Sudáfrica y el Reino Unido, lo que representa poblaciones geográfica y étnicamente diversas. El análisis incluyó 131 casos confirmados de COVID-19 detectados en dos regímenes de dosificación diferentes, incluidos 11,636 participantes reclutados en el brazo inglés y brasileño del estudio. El primer régimen se aplicó dos dosis completas con 4 semanas de diferencia en 8895 participantes adultos y mostró una eficacia del 62,1%. El segundo régimen, que se reconoció como el resultado de un error logístico, implicó la mitad de la primera dosis seguida de una dosis de refuerzo completa con la misma separación cronológica entre ellos e incluyó a 2741 individuos de 18 a 55 años que mostraban un 90 % eficacia. Se plantea la hipótesis de que esta discrepancia en la eficacia podría deberse a la combinación del grupo de edad más joven de la cohorte más pequeña y al hecho de que una dosis inicial más alta podría promover la inducción de anticuerpos contra el vector viral, dificultando así la intensidad de las respuestas inmunitarias inducidas por la dosis de refuerzo. La eficacia combinada de toda la cohorte de participantes 14 días después de la segunda dosis fue del 70,4% sin que se informaran casos graves u hospitalizaciones entre los participantes vacunados. Además, 3 semanas después de la administración de la primera dosis se reportaron diez casos de hospitalización por COVID-19, todos en el grupo control. Los datos preliminares sugieren que AZD1222 podría reducir la transmisión del virus ya que se observó una reducción en las infecciones asintomáticas. En el brazo de eficacia, se informa que el perfil de seguridad de la vacuna candidata es generalmente bueno y que las reacciones adversas son menos intensas y frecuentes en los participantes inmunizados de mayor edad que recibieron dosis más bajas y estos eventos disminuyen después de la segunda dosis.
AstraZeneca, con el apoyo de la Universidad de Oxford, presentó la seguridad y eficacia de la Fase 3 provisional completa a varios reguladores, incluidos el Reino Unido, la EMA y Brasil para su revisión y aprobación de uso de emergencia de su candidato. El 27 de noviembre, la MHRA emitió un comunicado de prensa informando que el Departamento de Salud y Asistencia Social del Reino Unido solicitó oficialmente la revisión de la vacuna candidata AZD1222 y el 30 de diciembre recibió la EUA para personas mayores de 18 años en el Reino Unido y Argentina. El 3 de enero de 2021, India otorgó la EUA a AZD1222, mientras que el 4 de enero comenzaron las primeras vacunas con AZD1222 en el Hospital Churchill de Oxford.
Gam-COVID-Vac / Sputnik V (Instituto de Investigación Gamaleya / Ministerio de Salud de la Federación de Rusia / Investigación y desarrollo de fármacos contractuales de Acellena): Los científicos del Instituto de Investigación Ruso Gamaleya desarrollaron la única vacuna candidata heteróloga de refuerzo primario del SARSCoV-2 hasta el momento con el fin de sortear el desafío de la inmunogenicidad reducida debido a los anticuerpos producidos contra el vector viral después de la primera inmunización. El serotipo del vector adenoviral utilizado para la vacunación de cebado es diferente del serotipo adenoviral utilizado como refuerzo. Por lo tanto, se seleccionó Ad26 con replicación defectuosa para entregar la información genética de la proteína Spike durante la primera vacunación y Ad5 recombinante con replicación defectuosa durante la segunda.
La vacuna candidata, recientemente rebautizada como Sputnik V, se probó en dos ensayos de Fase 1/2 a pequeña escala en los que participaron 38 participantes cada uno. Los resultados de los dos estudios se publicaron el 4 de septiembre, 3 semanas después de que el presidente Putin anunciara la autorización del Sputnik V para un uso limitado. El informe del ensayo de Fase 1/2 describió un buen perfil de seguridad con efectos adversos leves en una parte de los participantes vacunados, como astenia, mialgia, artralgia, fiebre, dolor de cabeza y dolor en el lugar de la inyección. El perfil inmunogénico del candidato a vacuna también fue bueno al inducir fuertes respuestas humorales en todos los participantes, así como la activación de las células T CD4 + y CD8 +.
El hecho de que el Ministerio de Salud de la Federación de Rusia haya aprobado Sputnik V como la primera vacuna para COVID-19 antes de los ensayos de seguridad y eficacia de la Fase 3 ha causado controversia y preocupación en la comunidad científica. Los ensayos de Fase 3 se planearon inicialmente para solo 2000 voluntarios, pero luego se reprogramaron para inscribir a 40,000 personas en 45 centros médicos diferentes en Rusia (Identificador de ensayo clínico: NCT04530396) y la República de Bielorrusia (Identificador de ensayo clínico: NCT04564716). El criterio de valoración principal es demostrar que Sputnik V evita que los participantes vacunados desarrollen COVID-19.
El 24 de noviembre, los desarrolladores de Sputnik V anunciaron los resultados de su segundo informe provisional de Fase 3 que reveló una eficacia del 91,4% de esta vacuna candidata después de analizar los datos obtenidos de 18.794 personas una semana después de recibir ambas dosis del régimen de inmunización heterólogo. Este análisis se basa en 39 casos confirmados de COVID-19 entre los participantes, de los cuales se informó que 8 pertenecían al grupo vacunado y 31 al grupo placebo. Según el comunicado de prensa, no se detectaron eventos adversos potencialmente mortales (Grado 4), mientras que los eventos graves más comunes (Grado 3) reportados fueron dolor en el lugar de la inyección y síntomas similares a los de la gripe como fiebre, fatiga y dolor de cabeza.
El 14 de diciembre, el Centro Nacional de Gamaleya y uno de sus patrocinadores, el Fondo Ruso de Inversión Directa (RDIF), anunciaron los resultados de su análisis intermedio final luego de llegar al punto de control de 78 casos confirmados de COVID-19. Se informó que la eficacia de Sputnik V tres semanas después de la administración de la primera dosis fue del 91,4% según el análisis de los datos obtenidos de 22.714 participantes (17.032 recibieron la vacuna y 5682). En el brazo inmunizado se notificaron 16 casos de COVID-19 frente a 62 casos en el brazo de placebo. Además, el anuncio informó 20 casos graves de COVID-19 en el grupo de placebo y ningún caso de enfermedad grave en el grupo vacunado, afirmando que la vacuna demostró una eficacia del 100% contra COVID-19 grave. Además de la aprobación de uso temprano de Sputnik V otorgada en Rusia, también se han emitido la EUA para esta vacuna desde Bielorrusia y Argentina.
JNJ-78436735 / Ad26.COV2.S (Centro Médico Janssen y Beth Israel Deaconess): Janssen Pharmaceuticals es la rama de desarrollo de vacunas de Johnson & Johnson Pharmaceutic. Su candidato es un vector basado en adenovirus 26 con replicación defectuosa que expresa la proteína S estabilizada previa a la fusión del SARS-CoV-2, un método desarrollado hace una década por investigadores del Beth Israel Deaconess Medical Center en Boston. Su principal diferencia con la vacuna candidata CanSino es el serotipo de adenovirus. A diferencia del omnipresente serotipo Ad5, muy pocas personas han estado expuestas al raro serotipo Ad26, por lo tanto, no se espera que la inmunidad preexistente contra el vector que reduce la inmunogenicidad de este candidato sea una preocupación importante. La segunda ventaja de este candidato es que el esquema de dosificación implica una única inmunización.
Janssen lanzó en julio un ensayo multicéntrico de Fase 1/2, aleatorizado, doble ciego y controlado con placebo, cuyos resultados se informaron recientemente. JNJ-78436735 se administró a niveles de dosis de 0,5 × 10(11) o 1 × 10(11) partículas virales por vacunación en participantes de dos grupos de edad diferentes: el primer grupo estaba compuesto por 402 adultos sanos de entre 18 y 55 años y el segundo grupo estaba compuesto por 394 ancianos sanos de 65 años o más.
La reactogenicidad de la vacuna fue leve y causó principalmente dolor en el lugar de la inyección, fiebre, dolor de cabeza y mialgia. Se detectaron anticuerpos específicos contra la proteína S en el 92% de los participantes del grupo más joven que recibieron cualquiera de las dosis y alcanzaron una tasa de seroconversión del 100% en el grupo de mayor edad. Se observaron respuestas de células T CD4 + en más del 80% de los miembros de cualquier grupo y también se documentaron respuestas robustas de células T CD8 +.
Estos resultados llevaron a un ensayo de Fase 3 que reclutó hasta 60.000 participantes que recibirán el nivel de dosis de 0,5 × 10(11) partículas virales (Identificador de ensayo clínico: NCT04505722). Los resultados primarios del ensayo analizados fueron la aparición de COVID-19 de moderado a grave. El 12 de octubre, los ensayos clínicos de Fase 3 de Janssen se detuvieron después de que un participante del estudio manifestara una reacción adversa seria (SAE). No se han publicado datos sobre cuál era la naturaleza de la enfermedad o si el participante había recibido la vacuna candidata o el placebo, pero el 23 de octubre, Jannsen anunció que el ensayo de Fase 3 de su candidata estaba a punto de reanudarse después de la recomendación de la Junta de Monitoreo y Seguridad de Datos (DSMB) que supervisa el estudio.
El 15 de noviembre, Janssen informó que iniciarán un segundo ensayo clínico de Fase 3, aleatorizado, doble ciego y controlado con placebo que estudia la seguridad y eficacia de un régimen de dos dosis de su candidato (Identificador de ensayo clínico: NCT04614948). En el estudio participarán 30.000 adultos de Bélgica, Colombia, Francia, Alemania, Filipinas, Sudáfrica, España, Reino Unido y Estados Unidos que recibirán dos dosis de la vacuna candidata Ad26.COV2.S o un placebo con un intervalo de 57 días.
Vacunas de patógenos inactivados
CoronaVac (Sinovac Research and Development Co.): Lanzada inicialmente con el nombre PiCoVacc, CoronaVac, es una vacuna candidata con adyuvante de alumbre de virus inactivado y purificado. El candidato fue producido por la activación de β-propiolactona de la cepa CN2 de SARS-CoV-2 aislada del lavado broncoalveolar de un paciente hospitalizado, esta cepa está estrechamente relacionada con la cepa 2019-nCoV-BetaCoV Wuhan / WIV04 / 2019. Las vacunas inactivadas presentan algunos desafíos técnicos como desventaja. El proceso de inactivación a veces puede dañar los antígenos y provocar una inmunogenicidad subóptima. Además, las vacunas inactivadas comúnmente necesitan varias dosis de refuerzo para producir respuestas inmunes fuertes y no activan tradicionalmente las respuestas celulares. Más importante aún, para mejorar su capacidad de provocar inmunidad, estas vacunas requieren la adición de adyuvantes. De hecho, CoronaVac es una vacuna candidata con adyuvante de alumbre.
Se llevó a cabo un ensayo de inmunogenicidad y seguridad de escalada de dosis, aleatorizado, doble ciego y controlado con placebo de Fase 2 en el que participaron 600 voluntarios sanos de entre 18 y 59 años que recibieron dos dosis diferentes de la vacuna (3 o 6 μg / 0,5 ml) o placebo. CoronaVac fue bien tolerado en ambas dosis y la mayoría de las reacciones adversas fueron leves. El dolor en el lugar de la inyección fue el síntoma más común de los reportados. Ambas dosis de CoronaVac indujeron una seroconversión en más del 90% de los individuos inmunizados, sin embargo, no se informaron respuestas de células T.
CoronaVac se encuentra actualmente en ensayos clínicos de Fase 3 en un régimen de inyección de dos dosis con un intervalo de 14 días. Estos ensayos inscribirán a 8870 participantes de Brasil (Identificador de ensayo clínico: NCT04456595), voluntarios de Indonesia (Número de registro: INA-WXFM0YX) y Turquía, para evaluar la eficacia de la vacuna en la prevención del COVID-19 y la frecuencia de efectos adversos.
Según se informa, CoronaVac recibió una aprobación de emergencia para uso limitado en julio en China, mientras que Sinovac está aumentando la producción de su vacuna candidata con el objetivo de suministrar a Indonesia 40 millones de dosis para marzo de 2021 y comenzar la distribución mundial a principios de 2021.
Nombre desconocido (Instituto de Productos Biológicos de Wuhan / Grupo Nacional de Biotecnología de China-Sinopharm): El Instituto de Productos Biológicos de Wuhan se asoció con la farmacéutica estatal Sinopharm para desarrollar una vacuna de virus inactivado y purificado que se envió a los ensayos clínicos de Fase 1 y Fase 2. Este candidato se desarrolló al aislar inicialmente la cepa WIV04 de SARS-CoV-2 de un paciente en el Hospital Jinyintan, Wuhan. A continuación, el virus se propagó en una línea celular Vero y se inactivó con β-propiolactona. Finalmente, la vacuna se sometió a un procedimiento de adsorción de adyuvante de alumbre.
El informe provisional de los ensayos de Fase 1 y Fase 2 sobre adultos sanos entre 18 y 59 años se publicó el 13 de agosto. En el ensayo de dosis y seguridad de Fase 1, 96 participantes recibieron una de tres dosis diferentes (2,5, 5 y 10 μg / dosis) o un control de adyuvante de alumbre solo en un régimen de tres inyecciones. En el ensayo de seguridad e inmunogenicidad de Fase 2, se asignó aleatoriamente a 224 adultos para recibir dos veces una inmunización de 5 μg / dosis con un intervalo de 2 o 3 semanas entre cada dosis o recibir el control de adyuvante solo. Los resultados mostraron que la vacuna candidata tenía un buen perfil de seguridad con solo efectos adversos leves documentados (principalmente dolor en el lugar de la inyección y fiebre), producía anticuerpos en voluntarios, algunos de los cuales experimentaron fiebre y otros efectos secundarios. Ambos grupos que recibieron la vacuna candidata produjeron concentraciones altas de anticuerpos neutralizantes contra el SARS-CoV-2, aunque fueron significativamente más altas en el grupo que recibió las dos inyecciones con 3 semanas de diferencia.
Actualmente se están llevando a cabo ensayos clínicos de Fase 3 en los Emiratos Árabes Unidos (número de registro: ChiCTR2000034780), Perú, Marruecos (número de registro: ChiCTR2000039000) y Bahrein.
BBIBP-CorV (Instituto de Biotecnología de Beijing / Grupo Nacional de Biotecnología de China-Sinopharm): El segundo candidato a vacuna de virus inactivado desarrollado por Sinopharm es el resultado de su colaboración con el Instituto de Productos Biológicos de Beijing. BBIBP-CorV se desarrolló mediante la inactivación mediada por β-propiolactona de la cepa SARSCoV-2 19nCoV-CDC-Tan-HB02 que se replicó en células Vero88 y se adyuvó con hidróxido de aluminio. El hidróxido de aluminio activa la subunidad del receptor NLRP3 del inflamasoma y promueve la secreción de altos niveles de IL-1β e IL-18 derivadas del inflamasoma, activando así los mecanismos proinflamatorios del sistema inmunológico.
En el estudio de Fase 1 de seguridad y aumento de dosis, 192 participantes recibieron 2 μg, 4 μg u 8 μg de la vacuna o el placebo. Se emplearon dos grupos de edad, a saber, 18 a 59 años y ≥ 60 años. El efecto adverso sistemático más común informado fue fiebre en menos del 10% de los candidatos. El perfil de seguridad de la vacuna fue bastante bueno ya que todas las reacciones adversas documentadas fueron leves o moderadas y no se informaron eventos graves. La inmunogenicidad del candidato fue mayor en el grupo de edad más joven (18-59 años) y las concentraciones de anticuerpos neutralizantes presentaron una inducción dependiente de la dosis.
En los ensayos de Fase 2, se reclutaron 448 voluntarios que recibieron una dosis de 8 μg o dos dosis de 4 μg de vacuna con 2, 3 o 4 semanas de diferencia. Nuevamente, las reacciones adversas notificadas fueron leves o moderadas y la reacción sistemática más frecuente fue fiebre en menos del 4% de los miembros de cada grupo de dosis. Las concentraciones de anticuerpos neutralizantes fueron significativamente más altas en los grupos que recibieron una inmunización de refuerzo con 4 μg / dosis y fueron más altas cuando las dos inmunizaciones estaban separadas por una distancia de 3 semanas. Como era de esperar, no se informaron respuestas inmunitarias celulares. En este momento BBIBP-CorV se encuentra en ensayos clínicos de Fase 3 en Argentina (Identificador de ensayo clínico: NCT04560881), Bahrein, Jordania, Egipto y Emiratos Árabes Unidos (Identificador de ensayo clínico: NCT04510207), (Número de registro: ChiCTR2000034780) que evalúa la incidencia de COVID-19 en personas que han recibido dos dosis de la vacuna.
Según los informes, las dos candidatas a vacunas de Sinopharm están programadas para estar listas para el mercado a finales de año. El 14 de septiembre, U.A.E. otorgó aprobación de emergencia para que las vacunas de Sinopharms se administraran a los trabajadores de la salud, antes de obtener datos a gran escala sobre su seguridad y eficacia. El 9 de diciembre, U.A.E. otorgó la aprobación total a BBIBP-CorV, seguida de Bahrein el 13 de diciembre. El 30 de diciembre, Sinopharm anunció que su candidato tenía una eficacia del 79,34% y recibió la aprobación en China al día siguiente. Por otro lado, Egipto emitió una autorización de emergencia el 3 de enero de 2021.
Covaxin / BBV152 (Bharat Biotech / Consejo Indio de Investigación Médica / Instituto Nacional de Virología): Bharat Biotech, con sede en India, y el Consejo Indio de Investigación Médica desarrollaron un candidato a vacuna de virión completo inactivado purificado llamado Covaxin. La vacuna se desarrolló mediante la inactivación con β-propiolactona de una cepa india del nuevo coronavirus aislada por el Instituto Nacional de Virología de la India y propagada en células Vero CCL-81.
El 15 de diciembre de 1999 se publicó una preimpresión de un ensayo clínico de Fase 1 de BBV152. Los autores informan los resultados obtenidos de 375 participantes que recibieron tres formulaciones diferentes de BBV152 (n = 100 por cada formulación) o el adyuvante Algel (a base de aluminio) (n = 75). Las tres formulaciones diferentes incluían 3 o 6 μg de SARS-CoV-2 inactivado con virión completo adsorbido en alumbre (Algel-imidazoquinolina) o 6 μg de SARSCoV-2 inactivado con virión completo adsorbido en Algel solo. Los participantes recibieron dos dosis intramusculares con 2 semanas de diferencia y se evaluó la seguridad e inmunogenicidad de cada formulación. Se encontró que los efectos adversos fueron leves o moderados con una tasa de incidencia entre el 10 y el 20% y el dolor en el lugar de la inyección fue el evento informado con mayor frecuencia. En ambos grupos de Algel-imidazoquinolina, se informó la inducción de concentraciones altas de anticuerpos anti-SARS-CoV-2, células T CD4 + y CD8 + y fueron significativamente más altas que en el grupo de solo Algel. Las respuestas de las células T parecían estar sesgadas por Th1, mientras que las tasas de seroconversión después de la segunda dosis fueron del 87,9% y el 91,9% para los grupos de 3 y 6 μg, respectivamente. Además, las concentraciones de anticuerpos neutralizantes contra el SARSCoV-2 y las tasas de seroconversión se evaluaron mediante un ensayo de microneutralización con SARS-CoV-2 y una prueba de neutralización por reducción de placa usando tres cepas diferentes para el desafío viral. Las concentraciones de anticuerpos neutralizantes fueron significativamente más altos en los dos grupos de Alum-imidazoquinolina que en el grupo de la vacuna con solo Algel, mientras que las tasas de seroconversión neutralizante fueron del 93,4% y el 86,4% en los grupos de 3 y 6 μg adyuvantes con Alum-imidazoquinolina, respectivamente, frente al 86,6% en el grupo de 6 μg solo de Algel.
Un estudio clínico de fase 3, aleatorizado, doble ciego, para evaluar la eficacia, seguridad e inmunogenicidad de este candidato comenzó el 23 de octubre (número de registro: CTRI / 2020/11/028976) en India.
Un total de 25.800 participantes adultos recibirán 3 μg de la forma con adyuvante del candidato o solución salina tamponada con fosfato con adyuvante como placebo, en un régimen de estimulación inicial de dos inmunizaciones intramusculares separadas por 4 semanas. La vacuna candidata contiene 6 μg de virus inactivado por vacunación. Los resultados iniciales del ensayo clínico de Fase 3 se esperan en el primer trimestre de 2021. El 3 de enero de 2021, India otorgó la EUA a BBV152, aunque todavía se reclutan participantes para los ensayos de eficacia y seguridad de Fase 3.
Vacunas de subunidades proteicas
NVX-CoV2373 (Novavax): De manera similar a las vacunas de patógenos inactivados, los candidatos a subunidades de proteínas generalmente exhiben un perfil de seguridad extremadamente favorable, pero requieren múltiples dosis de refuerzo y provocan respuestas celulares de bajo grado.
Novavax, con sede en Maryland, ha desarrollado una nanopartícula de glicoproteína recombinante SARS-CoV-2 S de prefusión de longitud completa expresada en un sistema de baculovirus-Sf9 y se administra con un adyuvante llamado Matrix M1. El adyuvante Matrix M1 basado en saponina se utiliza precisamente para abordar la ausencia de respuestas inmunes mediadas por células que caracterizan a las vacunas de subunidades proteicas.
Novavax lanzó un ensayo de Fase 1/2 cuyos resultados se publicaron en el New England Journal of Medicine el 2 de septiembre. Un total de 131 adultos sanos fueron asignados aleatoriamente para recibir dos administraciones de la vacuna con o sin el adyuvante o un placebo. Los efectos adversos producidos fueron nulos o leves y de corta duración. La adición de adyuvante mejoró las respuestas inmunes provocadas por el candidato a vacuna y dio como resultado respuestas celulares que exhibieron un fenotipo sesgado de Th1. Los anticuerpos anti-S IgG y neutralizantes inducidos por la vacunación superaron a los detectados en sueros convalecientes de pacientes con COVID-19.
El 23 de septiembre, Novavax lanzó una prueba de Fase 3 que tiene como objetivo inscribir hasta 9000 voluntarios en el Reino Unido (Número de registro: 2020-004123-16) y planea expandirse en Estados Unidos, India y otros países. Durante el mismo mes, Novavax estableció una colaboración con el Serum Institute of India que permitirá la producción de hasta 2 mil millones de dosis al año. El 28 de diciembre, Novavax inició un ensayo de Fase 3 en los EE. UU. con el objetivo de reclutar a 30.000 participantes, la mitad de los cuales recibirán 5 μg de nanopartícula de glicoproteína recombinante SARS-CoV-2 S de prefusión completa con 50 μg de adyuvante Matrix M1 (Identificador de ensayo clínico: NCT04611802).
De la lista de las diez principales vacunas candidatas, cinco (ARNm-1273, ARNm-BNT162b2, AZD1222, JNJ-78436735, NVX-CoV2373) forman parte de Operation Warp Speed, una iniciativa que se ha fijado el objetivo de entregar 300 millones de dosis de vacunas seguras y eficientes para mediados de 2021 en los EE. UU.
ZF2001 (Biofarmacéutica Anhui Zhifei Longcom / Academia China de Ciencias Médicas): El último candidato a vacuna de subunidad que ingresó en los estudios clínicos de Fase 3 es el antígeno dimérico RBD adyuvante diseñado por Anhui Zhifei Longcom Biopharmaceutical y el Instituto de Microbiología de la Academia China de Ciencias Médicas. El estudio clínico de fase 3 se lanzó en diciembre y se llevará a cabo inicialmente en China y Uzbekistán, mientras que Indonesia, Pakistán y Ecuador seguirán como sitios de estudio (Identificador de ensayo clínico: NCT04646590 y Número de registro: ChiCTR2000040153). El diseño del estudio implica el reclutamiento de 22.000 voluntarios de China y 7.000 sujetos fuera de China para un total de 29.000 voluntarios. Todavía no hay resultados publicados sobre este candidato; sin embargo, los datos de su ensayo clínico de Fase 2 controlado con placebo (Identificador de ensayo clínico: NCT04466085) realizado en un total de 900 participantes de entre 18 y 59 años sugieren que se evalúe un régimen de 2 o 3 dosis. Cada inmunización estará separada por la siguiente por 4 semanas.
Nombre desconocido (Sanofi Pasteur / GlaxoSmithKline): Este candidato está diseñado de manera similar a la vacuna tetravalente FluBlok producida por Sanofi. Sanofi utiliza un sistema de expresión de baculovirus para transferir la información genética del inmunógeno en células de insectos lepidópteros que posteriormente expresan altos niveles del antígeno codificado, en este caso, la proteína S del SARS-CoV-2. GSK está proporcionando su AS03 (Adjuvant System 3) adyuvante a base de escualeno que se ha utilizado con éxito en varias vacunas desarrolladas por GSK, como la vacuna contra la influenza pandémica A (H1N1) llamada Pandemrix. El 3 de septiembre, Sanofi anunció que entrarían en ensayos clínicos combinados de Fase 1/2 con su vacuna candidata. Este ensayo se realizó con la participación de 440 participantes en 11 sitios de investigación en los Estados Unidos (número de registro: NCT04537208). Los participantes se dividieron en tres grupos diferentes y recibieron una o dos dosis de la vacuna, o un control de placebo, respectivamente. Sin embargo, el 11 de diciembre Sanofi y GSK anunciaron que su candidato no logró obtener respuestas inmunes fuertes en participantes mayores de 50 años y solo mostró eficacia en adultos de 18 a 49 años. Así, las dos empresas planean optimizar la concentración de antígeno administrada por su candidato para mejorar su inmunogenicidad e iniciar una Fase 2b con una nueva formulación que se comparará con una vacuna SARS-CoV-2 ya autorizada.
Vacunas de partículas similares a virus
CoVLP (Medicago): Las vacunas de partículas similares a virus tienen como objetivo combinar la eficacia de las vacunas de patógenos atenuados con el excelente perfil de seguridad que generalmente se encuentra en las vacunas de subunidades. La VLP muestra múltiples copias del antígeno diana en su superficie y tiene un tamaño que favorece el reconocimiento y la absorción posterior de las células presentadoras de antígeno, lo que promueve su fagocitosis, procesamiento y presentación eficientes por las células dendríticas e induce fuertes respuestas adaptativas.
El único candidato avanzado contra el SARS-CoV-2 que emplea esta estrategia es la vacuna diseñada por la empresa Medicago con sede en Quebec. El enfoque de Medicago es único, ya que utiliza la planta transfectada con virus Nicotiana benthamiana para expresar la forma de la subunidad trimérica de prefusión de la proteína S del SARS-CoV-2 y ensamblarla en la superficie de las VLP que se cosechan y se usan para la inmunización.
Se llevó a cabo un estudio clínico de Fase 1 después de inscribir a 180 participantes, de 18 a 55 años de edad, que fueron sometidos a un régimen de dos vacunas de cualquiera de las tres dosis (3,75 μg, 7,5 μg y 15 μg) por dosis. Además, cada una de las dosis se complementó con adyuvantes CpG 1018 o AS03 o se aplicó sin adyuvante. CpG 1018 es un ligando del receptor 9 tipo Toll desarrollado por Dynavax que induce respuestas celulares y humorales robustas, mientras que el adyuvante AS03 basado en escualeno está desarrollado y patentado por GSK y se ha utilizado en varios de los productos de la empresa. Los resultados de estos estudios se publicaron en línea como una preimpresión el 6 de noviembre e indican que la administración de CoVLP fue bien tolerada ya que generó solo eventos adversos transitorios de leves a moderados. Además, no hubo efecto dosis-dependiente sobre la inducción de anticuerpos neutralizantes y la adición de ambos adyuvantes indujo fuertes respuestas humorales y celulares. Sin embargo, cuando se administró junto con AS03, incluso la dosis más baja de 3.75 μg de la vacuna candidata fue capaz de provocar fuertes respuestas de células T y niveles de anticuerpos neutralizantes que fueron aproximadamente 10 veces más altos que los producidos en promedio en individuos convalecientes por COVID-19. Con base en estos resultados, el 19 de noviembre comenzó el reclutamiento una Fase 2/3 aleatorizada, ciega al observador y controlada con placebo (Identificador de ensayo clínico: NCT04636697). El estudio incluirá a 30,612 voluntarios adultos que recibirán 2 dosis intramusculares de 3.75 μg de vacuna CoVLP con 0.5 mL de adyuvante AS03 o un placebo con una diferencia de 3 semanas.
Conclusiones
El mundo está en medio de una pandemia de COVID-19 y toda la comunidad científica de vacunología está corriendo para encontrar una vacuna contra el SARS-CoV-2 que sea segura y eficaz. Actualmente hay más de 230 candidatos a vacunas en desarrollo, y varios de ellos ya han recibido autorizaciones de uso de emergencia en menos de un año desde el primer informe de una infección por SARS-CoV-2.
Los comités de ética están revisando sus protocolos de autorización y las empresas farmacéuticas han formado alianzas estratégicas con la institución desarrolladora de vacunas para aumentar la producción de vacunas candidatas en riesgo. Más de 150 países han entrado en la iniciativa COVAX y otras alianzas que tendrán como objetivo asegurar una distribución equitativa de una vacuna aprobada.
Varios gobiernos han realizado pagos por adelantado para asegurar una serie de dosis de las vacunas en desarrollo que ayudarán a volver a la normalidad anterior a COVID-19.
El desarrollo de múltiples candidatos a vacunas que empleen diferentes sistemas de administración probablemente resultará crucial en la lucha para poner fin a la pandemia de COVID-19. Por un lado, varias opciones de vacunación, si se aprueban, permitirán producir las dosis necesarias para la vacunación masiva en un plazo más corto. Por otro lado, es muy posible que diferentes plataformas de vacunas exhiban diferentes grados de protección contra grupos de población específicos con respuestas inmunes alteradas, como niños, mujeres embarazadas, poblaciones inmunodeprimidas debido a comorbilidades y grupos de edad inmunosenescente ≥65 años.
Varios aspectos críticos de la inmunidad al SARS-CoV-2 serán esclarecidos como resultado de campañas masivas de vacunación. La durabilidad de la inmunidad inducida por las diferentes estrategias de vacuna, así como los detalles finos de las respuestas inmunes provocadas, surgirán a medida que se vacunen poblaciones más grandes, incluidas las personas con inmunidad subóptima.
Referencias:
Kyriakidis NC, López-Cortés A, González EV, Grimaldos AB, Prado EO. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines. 2021 Feb 22;6(1):28. doi: 10.1038/s41541-021-00292-w. PMID: 33619260.
Tomado de: https://www.nature.com/articles/s41541-021-00292-w
Nota VACUNAS

Vacuna BNT162b2 ARNm Covid-19 en un entorno de vacunación masiva a nivel nacional
Resumen:
ANTECEDENTES
A medida que comienzan las campañas de vacunación masiva contra la enfermedad del coronavirus 2019 (Covid-19) en todo el mundo, la efectividad de la vacuna debe evaluarse para una variedad de resultados en diversas poblaciones en un entorno no controlado. En este estudio, se utilizaron datos de la organización de atención médica más grande de Israel para evaluar la efectividad de la vacuna de ARNm BNT162b2.
MÉTODOS
Todas las personas que fueron vacunadas durante el período comprendido entre el 20 de diciembre de 2020 y el 1 de febrero de 2021 se emparejaron con controles no vacunados en una proporción de 1: 1 según las características demográficas y clínicas. Los resultados del estudio incluyeron infección documentada con el SARS-CoV-2, Covid-19 sintomático, hospitalización relacionada con Covid-19, enfermedad grave y muerte. Estimamos la efectividad de la vacuna para cada resultado como uno menos la razón de riesgo, utilizando el estimador de Kaplan-Meier.
RESULTADOS
Cada grupo de estudio incluyó a 596,618 personas. La efectividad estimada de la vacuna para los resultados del estudio en los días 14 a 20 después de la primera dosis y 7 o más días después de la segunda dosis fue la siguiente:
para infección documentada, 46% (intervalo de confianza [IC] del 95%, 40 a 51) y 92% (IC del 95%, 88 a 95);
para Covid-19 sintomático, 57% (IC del 95%, 50 a 63) y 94% (IC del 95%, 87 a 98);
para hospitalización, 74% (IC del 95%, 56 a 86) y 87% (IC del 95%, 55 a 100);
para la enfermedad grave, 62% (IC del 95%, 39 a 80) y 92% (IC del 95%, 75 a 100), respectivamente.
La efectividad estimada para prevenir la muerte por Covid-19 fue del 72% (IC del 95%, 19 a 100) durante los días 14 a 20 después de la primera dosis. La efectividad estimada en subpoblaciones específicas evaluadas para infección documentada y Covid-19 sintomático fue consistente en todos los grupos de edad, con una efectividad ligeramente menor en personas con múltiples afecciones coexistentes.
CONCLUSIONES
Este estudio en un entorno de vacunación masiva a nivel nacional sugiere que la vacuna de ARNm BNT162b2 es eficaz para una amplia gama de resultados relacionados con Covid-19, un hallazgo consistente con el del ensayo aleatorizado.
Introducción:
En muchas partes del mundo se están iniciando campañas de vacunación con vacunas recientemente aprobadas contra SARS-CoV-2. Los ensayos clínicos aleatorios de vacunas basadas en ARNm informaron eficacia para prevenir el coronavirus 2019 (Covid-19) en el rango de 94% a 95% .
Aunque los ensayos clínicos aleatorios se consideran el «estándar de oro» para evaluar los efectos de la intervención, tienen limitaciones notables de tamaño de muestra y análisis de subgrupos, criterios de inclusión restrictivos y un entorno altamente controlado que puede no replicarse en un lanzamiento masivo de vacunas. Por ejemplo, el ensayo de fase 3 de la vacuna de ARNm BNT162b2 contra Covid-19 incluyó a 21.720 personas que fueron asignadas al azar al grupo vacunado, lo que permitió estimaciones de la eficacia de la vacuna en solo un pequeño número de subpoblaciones. Además, los pacientes con enfermedades crónicas fueron incluidos sólo si los investigadores consideraron estables las condiciones médicas.
También es importante ver si en un programa de vacunación ampliado factores como el cumplimiento subóptimo de los programas de vacunación y la logística de manipulación de la vacuna influyen en la eficacia de la misma. Por lo tanto, los análisis posteriores a la autorización pueden satisfacer la necesidad urgente de evaluar la efectividad de las vacunas Covid-19 en diversas poblaciones con una amplia gama de condiciones coexistentes, en medio de una adherencia imperfecta a los protocolos de vacunación y los desafíos del mantenimiento de la cadena de frío y la logística de implementación de vacunas.
Aprovechamos los repositorios de datos integrados de la organización de atención médica más grande de Israel para evaluar la efectividad de la vacuna Covid-19 para cinco resultados:
- infección documentada por SARS-CoV-2,
- Covid-19 sintomático,
- hospitalización,
- enfermedad grave y
- muerte.
Utilizando este conjunto de datos de observación, evaluamos la efectividad a lo largo del tiempo y en subpoblaciones definidas por edad, sexo y condiciones coexistentes.
Métodos
POBLACIÓN DE ESTUDIO
Analizamos datos de Clalit Health Services (CHS), la más grande de las cuatro organizaciones integradas de atención médica en Israel, que asegura a 4,7 millones de pacientes (53% de la población). En el Apéndice complementario (Métodos complementarios 1) se proporciona una descripción de los repositorios de datos de CHS utilizados para este estudio, disponible con el texto completo de este artículo en NEJM.org. La información sobre el acceso de los autores a estos repositorios, así como las contribuciones de los autores al estudio, se proporciona en Métodos complementarios 2. Este estudio fue aprobado por la junta de revisión institucional de CHS. El estudio estuvo exento del requisito de consentimiento informado.
DISEÑO DEL ESTUDIO
Diseñamos este estudio observacional para emular un ensayo objetivo del efecto causal de la vacuna BNT162b2 en los resultados de Covid-19. Los criterios de elegibilidad incluyeron una edad de 16 años o más, sin tener un RT-PCR SARS-CoV-2 positivo documentado previamente, y ser miembro de la organización sanitaria durante los 12 meses anteriores.
Fueron excluidos los grupos de población en los que la variabilidad interna en la probabilidad de exposición o en los resultados es alta y no es factible controlar la alta variabilidad (p. ej., alta variabilidad en el riesgo de infección entre los trabajadores de la salud que atienden a los pacientes en salas dedicadas al Covid-19 en comparación con el personal administrativo). Dichos grupos de población son personas que no tienen un área de residencia geoestadística documentada, aquellas personas que han tenido interacciones con el sistema de atención médica durante los 3 días anteriores que pueden indicar el inicio de una enfermedad sintomática y pueden impedir la vacunación, residentes de hogares de ancianos, personas médicamente confinadas al hogar y trabajadores de la salud.
Cada día durante el período comprendido entre el 20 de diciembre de 2020 y el 1 de febrero de 2021, todas las personas recién vacunadas se emparejaron en una proporción de 1: 1 con los controles no vacunados. Para cada persona, el seguimiento finalizó al ocurrir alguno de los siguientes eventos: ocurrencia de un evento previsto como resultado, muerte no relacionada con Covid-19, vacunación (para controles no vacunados), vacunación del control emparejado (para personas vacunadas) o al final del período de estudio. Las personas recién vacunadas eran elegibles para su inclusión en el estudio, incluso si habían sido seleccionadas previamente como control.
Emparejamos a los receptores de la vacuna y a los controles en las variables asociadas con la probabilidad de vacunación y de infección o gravedad de Covid-19: edad, sexo, sector (judío general, árabe o judío ultraortodoxo), barrio de residencia (debido a que la actividad de la enfermedad y la captación de vacunación varía mucho entre áreas geoestadísticas definidas), antecedentes de vacunación contra la influenza durante los 5 años anteriores (0, 1 ó 2, 3 ó 4, ó ≥5 vacunas), embarazo (un factor de riesgo potencial para Covid-19 grave y asociado con la tasa de vacunación debido a los cambios en las pautas de vacunación para mujeres embarazadas) y el número total de afecciones coexistentes que los Centros para el Control y la Prevención de Enfermedades (CDC) habían identificado como factores de riesgo de Covid-19 grave al 20 de diciembre de 2020.(Consulte los métodos complementarios 3 para obtener información adicional)
El protocolo y el plan de análisis estadístico están disponibles en NEJM.org.
Los cinco resultados de interés fueron: infección documentada por SARS-CoV-2 confirmada por prueba de PCR positiva, Covid-19 sintomático documentado, ingreso hospitalario por Covid-19, Covid-19 grave (según los criterios de los Institutos Nacionales de Salud) 8 y muerte por Covid -19. Cada uno de estos resultados incluye los resultados que le siguen. En un análisis complementario, también evaluamos un resultado adicional, la infección por SARS-CoV-2 sin síntomas documentados, como una representación imperfecta de la infección asintomática (ya que los síntomas leves pueden no estar documentados).
ANÁLISIS ESTADÍSTICO
El balance de covariables después del emparejamiento se evaluó mediante el uso de una gráfica de las diferencias medias entre los valores de las variables (estandarizadas para las variables continuas) para los grupos vacunados y no vacunados, considerando aceptable una diferencia de 0,1 o menos. Curvas de supervivencia para los vacunados y los grupos no vacunados se estimaron con el estimador de Kaplan-Meier.
Consideramos tres períodos: días 14 a 20 después de la primera dosis de vacuna, días 21 a 27 después de la primera dosis (la administración de la segunda dosis estaba programada para el día 21 después de la primera dosis), y el día 7 después de la segunda dosis hasta el final del seguimiento. Para cada período, utilizamos el estimador de Kaplan-Meier con resultado diario y eventos de censura para calcular la probabilidad («riesgo») del resultado durante el período, utilizando pares emparejados en los que ambas personas todavía estaban en riesgo al comienzo del período. . Luego calculamos los cocientes de riesgo para la vacunación en comparación con ninguna vacuna y estimamos la efectividad de la vacuna como uno menos el cociente de riesgo. Estimamos la efectividad de la vacuna solo en análisis en los que hubo más de 10 casos de un resultado en los dos grupos.
El período inmediatamente posterior a la primera dosis, cuando la inmunidad se está construyendo gradualmente, se excluyó de los análisis principales porque se espera que la razón de riesgo sea cercana a 1 durante este período. En los análisis secundarios, consideramos los períodos desde el día 0 hasta el día 20 y desde el día 0 hasta el día 27, para evitar un posible sesgo de selección en los análisis principales que estaban restringidos a personas cuyos datos permanecieron sin censura al comienzo de cada período (ver Métodos complementarios 4) .11-13 También realizamos un análisis de sensibilidad en los 6 días posteriores a la segunda dosis de vacuna entre los que recibieron una segunda dosis. Un análisis de sensibilidad adicional estimó la razón de riesgo cada día para el resultado documentado de la infección por SARS-CoV-2.
Realizamos un análisis de sensibilidad adicional para evaluar el potencial de sesgo de selección debido a la censura informativa. En este análisis, los datos sobre los controles que se vacunaron posteriormente se censuraron solo después de 7 días (es decir, después del período con poco o ningún efecto de la vacuna) más el tiempo medio desde el diagnóstico documentado de Covid-19 hasta el resultado que se está estudiando (en estadística aplicada en investigaciones médicas la censura es el fenómeno que ocurre cuando el valor de una observación sólo se conoce parcialmente).
Calculamos los intervalos de confianza del 95% utilizando el método de arranque porcentual con 500 repeticiones. Los análisis se realizaron con el uso del software R, versión 4.0.2.
Resultados
POBLACIÓN DE ESTUDIO
De 1,503,216 miembros de CHS que fueron vacunados, 1,163,534 fueron elegibles para el estudio y 596,618 fueron emparejados con controles no vacunados (ver Figura 1 en el texto original). Las personas emparejadas eran más jóvenes que la población elegible en general y tenían una menor prevalencia de enfermedades crónicas porque había un grupo más pequeño de posibles coincidencias no vacunados para los receptores de vacunas de mayor edad, debido a las altas tasas de vacunación en la población de mayor edad. Las características basales de las personas emparejadas se muestran en la Tabla 1. Todas las variables estaban bien equilibradas entre los grupos de estudio. Aproximadamente el 0,6% de las personas a las que les faltaban datos sobre el tabaquismo o el índice de masa corporal se excluyeron del análisis (Figura 1). Los datos para el 44% de los controles no vacunados y sus pares emparejados fueron censurados cuando los controles recibieron la vacuna.
EFECTIVIDAD DE LA VACUNA
Durante un seguimiento medio de 15 días (rango intercuartílico, 5 a 25), se documentaron 10.561 infecciones (0,6 infecciones por 1.000 personas-día), de las cuales 5.996 (57%) fueron enfermedad sintomática de Covid-19, 369 requirieron hospitalización, 229 fueron casos graves de Covid-19 y 41 resultaron en la muerte. Las hospitalizaciones, la enfermedad grave y la muerte ocurrieron en períodos de tiempo cada vez mayores desde el diagnóstico (tiempos medios, 1, 5 y 11 días, respectivamente; ver Fig. S3). De las personas que tuvieron 21 o más días de seguimiento, el 96% recibió una segunda dosis de vacuna (el 95% de las cuales la recibió antes del día 24).
La Figura 2 muestra las curvas de incidencia acumulada para los resultados incluidos, y la Tabla 2 muestra la efectividad estimada de la vacuna para los principales resultados y períodos de tiempo. Durante el período de 14 a 20 días después de la primera dosis, la efectividad estimada de la vacuna para la infección documentada fue del 46% (intervalo de confianza [IC] del 95%, 40 a 51); enfermedad sintomática por Covid-19, 57% (IC del 95%, 50 a 63); hospitalización, 74% (IC del 95%, 56 a 86); enfermedad grave, 62% (IC del 95%, 39 a 80); y muerte, 72% (IC 95%, 19 a 100).
Durante el período de 21 a 27 días después de la primera dosis, la efectividad estimada para estos resultados fue del 60% (IC del 95%, 53 a 66), 66% (IC del 95%, 57 a 73), 78% (IC del 95% , 61 a 91), 80% (IC del 95%, 59 a 94) y 84% (IC del 95%, 44 a 100), respectivamente. En el período de seguimiento que comenzó 7 días después de la segunda dosis, la efectividad de la vacuna para infecciones documentadas, enfermedad sintomática, hospitalización y enfermedad grave fue del 92% (IC del 95%, 88 a 95), 94% (IC del 95%, 87 a 98), 87% (IC del 95%, 55 a 100) y 92% (IC del 95%, 75 a 100), respectivamente. El valor diario de uno menos la razón de riesgo para el resultado de la infección documentada se incluye en la Figura S4; es consistente con un aumento diario gradual de la eficacia de la vacuna.
La Tabla 3 muestra la efectividad estimada de la vacuna para la infección documentada por SARS-CoV-2 y los resultados de Covid-19 en subpoblaciones definidas por edad, sexo y condiciones coexistentes. Las estimaciones son consistentes con una efectividad similar en todos los grupos de edad y una efectividad ligeramente menor entre los pacientes con múltiples afecciones coexistentes.
La efectividad estimada de la vacuna para el proxy de infección asintomática fue del 29% (IC del 95%, 17 a 39) durante el período de 14 a 20 días después de la primera dosis, 52% (IC del 95%, 41 a 60) 21 a 27 días después la primera dosis y el 90% (IC del 95%, 83 a 94) 7 o más días después de la segunda dosis.
La Figura S6 muestra una ampliación de la curva de incidencia acumulada para el resultado de la enfermedad sintomática, mostrando la divergencia de las curvas que comienzan alrededor del día 12. Esto se muestra en comparación con la misma curva de un análisis mínimamente emparejado (solo por edad y sexo) que muestra una separación más temprana y más amplia de las curvas.
La Tabla S4 muestra los análisis de sensibilidad de la efectividad de la vacuna durante períodos de seguimiento adicionales. Las estimaciones de efectividad acumulada a partir del día 0 fueron menores en todos los resultados. Las estimaciones de eficacia condicionadas a la recepción de la segunda dosis de vacuna fueron más altas que las estimaciones incondicionales para los días 21 a 27 después de la primera dosis.
La Tabla S5 y la Figura S7 muestran los resultados del análisis de sensibilidad en el que los datos de las personas que se inscribieron como controles y luego se vacunaron se censuraron con retraso (varios días después de la fecha de vacunación, según el resultado). Las estimaciones son similares a las del análisis principal. La Tabla S6 detalla todos los análisis realizados durante el estudio, y la Tabla S7 incluye las tablas de vida para los diversos resultados.
Discusión
Este estudio evalúa la efectividad de la nueva vacuna de ARNm BNT162b2 contra Covid-19 en un entorno de vacunación masiva a nivel nacional. La efectividad estimada de la vacuna durante el período de seguimiento que comenzó 7 días después de la segunda dosis fue del 92% para la infección documentada, el 94% para el Covid-19 sintomático, el 87% para la hospitalización y el 92% para el Covid-19 grave. La efectividad estimada durante los días 14 a 20 (después de una dosis) y los días 21 a 27 (cambio gradual entre la primera y la segunda dosis de la vacuna) fue del 46% y 60% para la infección documentada, 57% y 66% para Covid-19 sintomático, 74 % y 78% por hospitalización, 62% y 80% por Covid-19 grave y 72% y 84% por muerte relacionada con Covid-19, respectivamente.
El primer criterio de valoración principal evaluado en el ensayo aleatorizado de la vacuna BNT162b2 fue Covid-19 sintomático. Tanto en el ensayo aleatorizado como en nuestro estudio, la incidencia acumulada de Covid-19 sintomático en los grupos vacunados y no vacunados comenzó a divergir alrededor del día 12 después de la primera dosis. La eficacia estimada de la vacuna para Covid-19 sintomático a partir del día 7 después de la la segunda dosis fue del 95% en el ensayo aleatorizado, en comparación con el 94% en nuestro estudio. La eficacia estimada entre la primera dosis y la segunda dosis fue del 52% en el ensayo, en comparación con el 29% en nuestro estudio. Esta diferencia puede reflejar el alto nivel de transmisión en Israel durante el período de estudio, que afectó tanto a las personas vacunadas como a los controles por igual durante los primeros 12 días después de la administración de la primera dosis. Para eliminar esta distorsión, estimamos la efectividad de la primera dosis de la vacuna contra Covid-19 para el período comprendido entre los días 14 y 20; la efectividad estimada fue del 57%.
La efectividad estimada para la infección documentada durante los días 14 a 20 fue del 46% en nuestro estudio. Chodick et al. informaron una efectividad relativamente similar del 51%, quienes evaluaron una cohorte de otra organización de atención médica en Israel y utilizaron un diseño de estudio diferente que comparó la infección entre personas vacunadas en los días 13 a 24 después de la primera dosis contra la infección.
En el ensayo aleatorizado, la eficacia estimada de la vacuna para Covid-19 grave (89% durante todo el período de estudio) se basó en sólo 10 casos. Nuestro estudio registró 229 casos de Covid-19 severo: 55 en el grupo vacunado y 174 en el grupo no vacunado, lo que resultó en una efectividad estimada del 62% para los días 14 a 20 después de la primera dosis, 80% para los días 21 a 27, y 92% durante 7 ó más días después de la segunda dosis.
El gran tamaño de la muestra en nuestro estudio también nos permitió estimar la efectividad de la vacuna para subpoblaciones específicas para las que el ensayo aleatorio no tenía el poder suficiente para evaluar. En el ensayo, la eficacia estimada de Covid-19 entre personas de hasta 55 años, mayores de 55 años y 65 años o más 7 días después de la segunda dosis fue de 94 a 96%. Pudimos estudiar grupos de edad más granulares y estimamos que la efectividad de la vacuna fue similar para los adultos de 70 años o más y para los grupos de edad más jóvenes durante el mismo período de tiempo.
El ensayo aleatorizado estimó la eficacia de la vacuna para pacientes con una o más condiciones coexistentes según el índice de comorbilidad de Charlson y específicamente para pacientes con obesidad o hipertensión. Estas medidas no brindan claridad con respecto a la efectividad en pacientes con múltiples afecciones coexistentes. Estimamos la efectividad de la vacuna en relación con varios números de condiciones coexistentes y encontramos indicios de que la efectividad puede ser ligeramente menor entre personas con un mayor número de condiciones coexistentes.
Dos factores hacen que el presente estudio sea especialmente adecuado para evaluar la efectividad de la vacuna BNT162b2 en una aplicación práctica: primero, una combinación poco común de ricos datos de antecedentes médicos, resultados de la prueba de PCR Covid-19 (para el resultado de infección documentado) y seguimiento del paciente tanto en la comunidad (para el resultado sintomático Covid-19) como en entornos de pacientes hospitalizados (para todos los demás resultados).
CHS ha mantenido un depósito de datos integrado para más de la mitad de la población israelí y lo ha actualizado diariamente durante más de dos décadas ; y segundo, el ritmo rápido y la alta aceptación de la vacuna Covid-19 en Israel y las altas tasas de enfermedad durante la campaña de vacunación. Por otro lado, el rápido ritmo de la campaña de vacunación contribuyó a la frecuente censura de los datos de los controles no vacunados emparejados, especialmente entre los mayores de 60 años (a menudo solo unos días después del emparejamiento) y la correspondiente reducción en el seguimiento promedio. -período inicial del estudio.
Han surgido preocupaciones con respecto a la posible resistencia de las variantes del SARS-CoV-2 a las vacunas Covid-19 y a los anticuerpos neutralizantes. Durante el período de estudio, una proporción cada vez mayor de aislamientos de SARS-CoV-2 en Israel, hasta un 80% en los días previos a la extracción de datos – eran de la variante B.1.1.7.21
Por lo tanto, este estudio estima una efectividad promedio de la vacuna en múltiples cepas. Aunque no podemos proporcionar una estimación de la eficacia específica para la variante B.1.1.7, la meseta observada durante los últimos períodos en la curva de incidencia acumulada para las personas vacunadas sugiere que la vacuna BNT162b2 también es eficaz para esta variante, una observación consistente con informes anteriores. que mostraban títulos de anticuerpos neutralizantes conservados. Se estimó que la variante B.1.351 era rara en Israel en el momento de la extracción de datos.
Al igual que con cualquier estudio observacional, nuestro estudio puede haber sido afectado por confusión residual debido a diferencias entre las personas vacunadas y los controles no vacunados, especialmente en términos de comportamiento de búsqueda de salud. Por lo tanto, realizamos un emparejamiento riguroso en una amplia gama de factores que pueden confundir el efecto causal de la vacuna en los diversos resultados. Después del proceso de emparejamiento, encontramos un patrón consistente de similitud entre los grupos en los días justo antes del día 12 después de la primera dosis (el inicio anticipado del efecto de la vacuna), que por lo tanto sirve como un período de «control negativo» . Esta similitud se produjo a pesar de un aumento temporal de eventos entre los controles no vacunados durante los primeros días después de la primera dosis de vacuna, probablemente debido al hecho de que las personas que optan por vacunarse en un día específico se sienten bien en el momento de la vacunación. La similitud de los grupos de estudio en condiciones coexistentes y factores de riesgo conocidos para Covid-19 grave proporciona más evidencia de intercambiabilidad (es decir, ausencia de confusión). Sin embargo, este riguroso proceso de emparejamiento tuvo el costo de no incluir en la cohorte final aproximadamente el 34% de las personas vacunadas que de otra manera cumplían con los criterios de elegibilidad del estudio. El emparejamiento limitado solo por edad y sexo hubiera sido insuficiente para eliminar el factor de confusión inicial.
También se excluyeron los grupos de población con alta variabilidad interna en la probabilidad de vacunación o resultado, como los trabajadores de la salud, las personas confinadas en el hogar por razones médicas y los residentes de hogares de ancianos, para evitar confusión residual. Aunque también fue menos probable que el ensayo aleatorizado incluyera a personas que no estaban lo suficientemente saludables para cumplir con las visitas programadas y el plan de vacunación, no excluyó a los trabajadores de la salud.
Para evaluar un posible sesgo de selección que podría derivarse de la censura informativa, por la cual los controles que están vacunados se sienten bien en el momento de la vacunación, realizamos un análisis de sensibilidad en el que se mantuvieron en el grupo no vacunado durante un período de tiempo que se estableció de manera diferente para cada resultado. Este análisis mostró resultados similares a los del análisis principal, lo que sugiere que dicho sesgo fue pequeño en nuestro análisis.
Finalmente, la fecha de inicio de los síntomas no estuvo disponible para el análisis. En cambio, para los resultados de la infección, la fecha se fijó en la fecha de recogida del hisopo para la primera prueba de PCR positiva. Dado que es probable que haya habido un lapso de tiempo entre el inicio de los síntomas y la recolección de los hisopos, la divergencia observada de las gráficas de incidencia acumulada para los resultados de la infección entre las personas vacunadas y los controles no vacunados puede retrasarse levemente. Paralelamente, podría haber una subestimación de la efectividad de la vacuna en cada ventana de tiempo, ya que la estimación en realidad refleja una ventana más estrecha para que la vacuna esté activa. Debido a que la prueba de PCR del SARS-CoV-2 es muy accesible en Israel y se puede realizar en cuestión de horas, estimamos que esta brecha de tiempo potencial y, por lo tanto, la subestimación de la efectividad de la vacuna es pequeña. Al interpretar las estimaciones de efectividad para los resultados más graves, se deben tener en cuenta las brechas medias más largas : 1 día para la hospitalización, 5 días para Covid-19 grave y 11 días para la muerte por Covid-19.
Este estudio estima una alta eficacia de la vacuna BNT162b2 para prevenir el Covid-19 sintomático en un entorno no controlado, similar a la eficacia de la vacuna informada en el ensayo aleatorizado. Nuestro estudio también sugiere que la efectividad es alta para los resultados más graves: hospitalización, enfermedad grave y muerte. Además, el beneficio estimado aumenta en magnitud a medida que pasa el tiempo. Estos resultados refuerzan la expectativa de que las vacunas recientemente aprobadas puedan ayudar a mitigar los profundos efectos globales de la pandemia Covid-19.
El Dr. Lipsitch recibe apoyo del Fondo Morris-Singer.
Los formularios de divulgación proporcionados por los autores están disponibles con el texto completo de este artículo en NEJM.org.
Drs. Dagan y Barda contribuyeron igualmente a este artículo.
Este artículo fue publicado el 24 de febrero de 2021 en NEJM.org.
Artículo original:
https://www.nejm.org/doi/full/10.1056/NEJMoa2101765
Protocolo: https://www.nejm.org/doi/suppl/10.1056/NEJMoa2101765/suppl_file/nejmoa2101765_protocol.pdf
Apéndice: https://www.nejm.org/doi/suppl/10.1056/NEJMoa2101765/suppl_file/nejmoa2101765_appendix.pdf
Declaraciones: https://www.nejm.org/doi/suppl/10.1056/NEJMoa2101765/suppl_file/nejmoa2101765_disclosures.pdf
Vacuna BNT162b2 ARNm Covid-19 en un entorno de vacunación masiva a nivel nacional


Correlación clínica de los perfiles de anticuerpos anti-SARS-CoV-2 en pacientes españoles con COVID-19 de una región de alta incidencia
El sello distintivo de las pruebas de laboratorio para el SARS-CoV-2 en casos agudos sospechosos de COVID-19 es la detección de ARN viral mediante la reacción en cadena de la polimerasa (rt-PCR). Con el tiempo, también se han puesto a disposición diferentes ensayos para la detección de anticuerpos contra diferentes antígenos diana del SARS-CoV-2. Si bien se asume ampliamente que los anticuerpos contra el SARS-CoV-2 se pueden usar como marcador de una infección pasada o presente por el SARS-CoV-2 y que son un marcador de inmunidad contra él, muchas preguntas que los rodean no han sido respondidas definitivamente:
- ¿Todos los pacientes con infecciones confirmadas por PCR con SARS-CoV-2 desarrollan anticuerpos y, de no ser así, qué significa eso para los pacientes que desarrollan o no anticuerpos? Un estudio reciente sugirió que hasta el 30% de los pacientes no desarrollan anticuerpos después de una infección confirmada por PCR con SARS-CoV-2.
- ¿El desarrollo de anticuerpos es útil para erradicar el virus o incluso causan daño a través de mecanismos inmunopatológicos? Han habido consideraciones recientes sobre la posibilidad de una mejoría clínica dependiente de anticuerpos de SARS-CoV-2
- ¿Se pueden utilizar anticuerpos contra el SARS-CoV-2 como marcador pronóstico? Y si es así, ¿mejoran o empeoran el pronóstico de un paciente?
Durante un brote epidémico ocurrido en octubre de 2020 en España, se examinaron los sueros de 347 pacientes con diagnóstico confirmado de COVID-19 por rt-PCR, con el objetivo de determinar: qué factores (sexo, edad, gravedad de la enfermedad) estarían relacionados con el desarrollo de anticuerpos contra el virus, y si la presencia de anticuerpos puede ser utilizada como un marcador de gravedad de la enfermedad.
Las pruebas de anticuerpos se realizaron para detectar la presencia de IgA e IgG contra la proteína Spike del SARS-CoV-2, mediante la técnica de ELISA.
Se tuvieron en cuenta datos demográficos de los pacientes como sexo y edad, así como la cantidad de días que transcurrieron entre el inicio de los síntomas y la recolección de la muestra de suero. La gravedad de la enfermedad se evalúo teniendo en cuenta sí el paciente requirió hospitalización, cuidados intensivos, asistencia respiratoria o falleció.
De los 347 pacientes estudiados, se detectó la presencia de anticuerpos IgA y/o IgG contra la proteína Spike del SARS-CoV-2 en 281, mientas que 65 pacientes fueron negativos.
Los resultados para la población en estudio demuestran que la presencia y, hasta cierto punto, los niveles de anticuerpos anti-SARS-CoV-2 dependen principalmente del tiempo transcurrido entre la aparición de los síntomas y la toma de la muestra de suero. Sin embargo, la presencia y el nivel de anticuerpos no se asoció con la edad, el sexo, ni la gravedad de la enfermedad. En este caso, se encontró que la edad avanzada fue principal factor de riesgo para un resultado adverso.
Referencia:
Markewitz, R., Torge, A., Wandinger, KP. et al. Clinical correlates of anti-SARS-CoV-2 antibody profiles in Spanish COVID-19 patients from a high incidence region. Sci Rep 11, 4363 (2021).
Más información en:
https://doi.org/10.1038/s41598-021-83969-5
Anticuerpos COVID-19
